10-K: Annual report pursuant to Section 13 and 15(d)
Published on March 1, 2022
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
(Mark One)
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the fiscal year ended December 31 , 2021
OR
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the transition period from to .
Commission file number 001-33528
(Exact Name of Registrant as Specified in Its Charter)
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) |
|||||||
| (Address of Principal Executive Offices) (Zip Code) | ||||||||
| (Registrant’s Telephone Number, Including Area Code) | ||||||||
| Securities registered pursuant to section 12(b) of the Act: | ||||||||
| Title of Each Class | Trading Symbol | Name of Each Exchange on Which Registered | ||||||
Securities registered pursuant to section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ý No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| ý | Accelerated filer | o | |||||||||
| Non-accelerated filer | o | Smaller reporting company | |||||||||
| Emerging growth company | |||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ý
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates computed by reference to the price at which the common equity was last sold, as of the last business day of the registrant’s most recently completed second fiscal quarter was: $1,607,181,353 .
As of February 25, 2022, the registrant had 681,455,306 shares of its common stock, par value $0.01 per share (“Common Stock”) outstanding.
Documents Incorporated by Reference
TABLE OF CONTENTS
| Page | |||||||||||
Part I. |
|||||||||||
| Item 1. | Business |
||||||||||
| Item 1A. | Risk Factors |
||||||||||
| Item 1B. | Unresolved Staff Comments |
||||||||||
| Item 2. | Properties |
||||||||||
| Item 3. | Legal Proceedings |
||||||||||
| Item 4. | Mine Safety Disclosures |
||||||||||
Part II. |
|||||||||||
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
||||||||||
| Item 6. | Selected Financial Data |
||||||||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
||||||||||
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
||||||||||
| Item 8. | Financial Statements and Supplementary Data |
||||||||||
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
||||||||||
| Item 9A. | Controls and Procedures |
||||||||||
| Item 9B. | Other Information |
||||||||||
| Item 9C. | Disclosure regarding Foreign Jurisdictions that Prevent Inspections. | ||||||||||
Part III. |
|||||||||||
| Item 10. | Directors, Executive Officers and Corporate Governance | ||||||||||
| Item 11. | Executive Compensation | ||||||||||
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | ||||||||||
| Item 13. | Certain Relationships and Related Transactions and Director Independence | ||||||||||
| Item 14. | Principal Accounting Fees and Services | ||||||||||
Part IV. |
|||||||||||
| Item 15. | Exhibits, Financial Statement Schedules |
||||||||||
| Item 16. | Form 10-K Summary | ||||||||||
Signatures |
|||||||||||
Certifications |
|||||||||||
| EX-21 | |||||||||||
| EX-23.1 | |||||||||||
| EX-31.1 | |||||||||||
| EX-31.2 | |||||||||||
| EX-32.1 | |||||||||||
| EX-32.2 | |||||||||||
| EX-101. INS XBRL Instance Document | |||||||||||
EX-101.SCH XBRL Taxonomy Extension Schema Document |
|||||||||||
EX-101.CAL XBRL Taxonomy Extension Calculation Linkbase Document |
|||||||||||
EX-101.DEF XBRL Taxonomy Extension Definition Linkbase Document |
|||||||||||
EX-101.LAB XBRL Taxonomy Extension Label Linkbase Document |
|||||||||||
EX-101.PRE XBRL Taxonomy Extension Presentation Linkbase Document |
|||||||||||
3
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements,” as that term is defined under the Private Securities Litigation Reform Act of 1995 (“PSLRA”), Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements include statements about our expectations, beliefs or intentions regarding our product development efforts, business, financial condition, results of operations, strategies or prospects, including the potential impact of the COVID-19 pandemic on our businesses, operating results, cash flows and/or financial condition. You can identify forward-looking statements by the fact that these statements do not relate strictly to historical or current matters. Rather, forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements. These factors include those described below and in “Item 1A-Risk Factors” of this Annual Report on Form 10-K. We do not undertake an obligation to update forward-looking statements. We intend that all forward-looking statements be subject to the safe-harbor provisions of the PSLRA. These forward-looking statements are only predictions and reflect our views as of the date they are made with respect to future events and financial performance.
Risks and uncertainties, the occurrence of which could adversely affect our business, include the following:
•our business may be materially adversely affected by the coronavirus (COVID-19) pandemic, including the impact on our sales and operations from continued or increasing infection rates and potential declines in testing needs should infection rates decline;
•we have had a history of losses and may not generate sustained positive cash flow sufficient to fund our operations and research and development programs;
•our need for, and ability to obtain, additional financing when needed on favorable terms, or at all;
•adverse results in material litigation matters or governmental inquiries;
•the risks inherent in developing, obtaining regulatory approvals for and commercializing new, commercially viable and competitive products and treatments;
•our research and development activities may not result in commercially viable products;
•that earlier clinical results of effectiveness and safety may not be reproducible or indicative of future results;
•that we may fail to obtain regulatory approval for Somatrogon (hGH-CTP) in the United States or successfully commercialize Rayaldee and Somatrogon (hGH-CTP);
•that we may not generate or sustain profits or cash flow from our laboratory operations or substantial revenue from Rayaldee and our other pharmaceutical and diagnostic products;
•that currently available over-the-counter and prescription products, as well as products under development by others, may prove to be as or more effective than our products for the indications being studied;
•our ability and our distribution and marketing partners’ ability to comply with regulatory requirements regarding the sales, marketing and manufacturing of our products and product candidates and the operation of our laboratories;
•the performance of our third-party distribution partners, licensees and manufacturers over which we have limited control;
•our success is dependent on the involvement and continued efforts of our Chairman and Chief Executive Officer;
•availability of insurance coverage with respect to material litigation matters;
•business combinations may disrupt our business, distract our management, may not proceed as planned, and may also increase the risk of potential third party claims and litigation;
•changes in regulation and policies in the United States (“U.S.”) and other countries, including increasing downward pressure on healthcare reimbursement;
•our ability to manage our growth and our expanded operations;
•increased competition, including price competition;
4
•changing relationships with payors, including the various state and multi-state programs, suppliers and strategic partners;
•efforts by third-party payors to reduce utilization and reimbursement for clinical testing services;
•our ability to maintain reimbursement coverage for our products and services, including Rayaldee and the 4Kscore test;
•failure to timely or accurately bill and collect for our services;
•the information technology systems that we rely on may be subject to unauthorized tampering, cyberattack or other data security or privacy incidents that could impact our billing processes or disrupt our operations;
•failure to obtain and retain new clients and business partners, or a reduction in tests ordered or specimens submitted by existing clients;
•failure to establish, and perform to, appropriate quality standards to assure that the highest level of quality is observed in the performance of our testing services;
•failure to maintain the security of patient-related information;
•our ability to obtain and maintain intellectual property protection for our products;
•our ability to defend our intellectual property rights with respect to our products;
•our ability to operate our business without infringing the intellectual property rights of others;
•our ability to attract and retain key scientific and management personnel;
•the risk that the carrying value of certain assets may exceed the fair value of the assets causing us to impair goodwill or other intangible assets;
•failure to obtain and maintain regulatory approval outside the U.S.; and
•legal, economic, political, regulatory, currency exchange, and other risks associated with international operations.
Risk Factor Summary
Our business is subject to numerous risks and uncertainties, including those described in Item 1A “Risk Factors”. These risks include, but are not limited to the following:
•Our business has been, and may continue to be, affected by the recent coronavirus disease 2019 (COVID-19) outbreak;
•We have had a history of operating losses and may not be able to sustain profitability in the near future;
•Our research and development activities may not result in commercially viable products;
•Our business is substantially dependent on our ability to generate profits and cash flow from our laboratory operations;
•Failure to timely or accurately bill and collect for our services could have a material adverse effect on our revenues and our business;
•The information technology systems that we rely on may be subject to unauthorized tampering, cyberattack or other data security incidents that could impact our billing processes or disrupt our operations;
•If we are unable to obtain and enforce patent protection for our products, our business could be materially harmed;
•Failure to maintain the security of patient-related information or compliance with security requirements could damage our reputation with customers, cause us to incur substantial additional costs and become subject to litigation;
•Failure to obtain regulatory approval within and outside the U.S. will prevent us from marketing our products and product candidates domestically and abroad;
•We are subject to risks associated with doing business globally; and
•Funding may not be available for us to continue to make acquisitions, investments and strategic alliances in order to grow our business.
5
PART I
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to the “Company”, “OPKO”, “we”, “our”, “ours”, and “us” refer to OPKO Health, Inc., a Delaware corporation, including our wholly-owned subsidiaries.
ITEM 1. BUSINESS
OVERVIEW
We are a diversified healthcare company that seeks to establish industry-leading positions in large and rapidly growing medical markets. Our diagnostics business includes BioReference Laboratories, Inc. (“BioReference”), one of the nation’s largest full service laboratories with an almost 250-person sales and marketing team to drive growth and leverage new products. Our pharmaceutical business features Rayaldee, a U.S. Food and Drug Administration (“FDA”) approved treatment for secondary hyperparathyroidism (“SHPT”) in adults with stage 3 or 4 chronic kidney disease (“CKD”) and vitamin D insufficiency, and a pipeline of products in various stages of development. Our leading product in development is Somatrogon (hGH-CTP), a once-weekly human growth hormone injection for which we completed a successful phase 3 study in August 2019 and is partnered with Pfizer Inc. (“Pfizer”). Regulatory applications for Somatrogon have been submitted to several countries around the world for review. In February 2022, the European Commission granted marketing authorization in the European Union for Somatrogon under the brand name NGENLA® to treat children and adolescents from as young as 3 years of age with growth disturbance due to insufficient secretion of growth hormone. In January 2022, the Ministry of Health, Labour and Welfare in Japan approved NGENLA® (Somatrogon) for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. In October 2021, Health Canada approved NGENLA® for the long-term treatment of pediatric patients who have growth hormone deficiency, and in November 2021, Australia’s Therapeutic Goods Administration approved NGENLA® for the long-term treatment of pediatric patients with growth disturbance due to insufficient secretion of growth hormone. We also submitted the initial Biologics License Application (“BLA”) with the FDA for approval of Somatrogon in the United States and Pfizer received a complete response letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine an appropriate path forward for the advancement of hGH (Somatrogon).
Through BioReference, we provide laboratory testing services, primarily to customers in the larger metropolitan areas in New York, New Jersey, Florida, Texas, Maryland, California, Pennsylvania, Delaware, Washington, DC, Illinois and Massachusetts, as well as to customers in a number of other states. We offer a comprehensive test menu of clinical diagnostics for blood, urine and tissue analysis. This includes hematology, clinical chemistry, immunoassay, infectious disease, serology, hormones, and toxicology assays, as well as Pap smear, anatomic pathology (biopsies) and other types of tissue analysis, as well as testing for COVID-19. We market our laboratory testing services directly to physicians, geneticists, hospitals, clinics, correctional and other health facilities.
We operate established pharmaceutical platforms in Spain, Ireland, Chile, and Mexico, which are generating revenue and from which we expect to generate positive cash flow and facilitate future market entry for our products currently in development. We have a development and commercial supply pharmaceutical company as well as a global supply chain operation and holding company in Ireland. We also own a specialty active pharmaceutical ingredients (“APIs”) manufacturer in Israel, which we expect will facilitate the development of our pipeline of molecules and compounds for our proprietary molecular diagnostic and therapeutic products.
We have a highly experienced management team. Based on the members’ respective experience in the industry, we believe that our management team has extensive development, regulatory and commercialization expertise and relationships that provide access to commercial opportunities.
All product or service marks appearing in type form different from that of the surrounding text are trademarks or service marks owned, licensed to, promoted or distributed by OPKO, its subsidiaries or affiliates, except as noted. All other trademarks or services marks are those of their respective owners.
GROWTH STRATEGY
We expect our future growth to come from leveraging our commercial infrastructure, proprietary technology and development strengths.
We launched our first pharmaceutical product, Rayaldee, in the U.S. market in the fourth quarter of 2016. We have under development a broad and diversified portfolio of diagnostic tests, small molecules, and biologics targeting a broad range of unmet medical needs. We also operate one of the largest full service laboratories in the U.S. We intend to continue to leverage
6
our proprietary technology and our strengths in all phases of research and development to further develop and commercialize our portfolio of proprietary pharmaceutical and diagnostic products. In support of our strategy, we intend to:
•continue to enhance our commercialization capability in the U.S. and internationally;
•develop and commercialize Rayaldee for new indications, including the treatment of SHPT in patients with vitamin D insufficiency and stage 5 CKD requiring regular hemodialysis;
•obtain requisite regulatory approval and compile clinical data for our most advanced product candidates; and
•expand into other medical markets that provide significant opportunities and that we believe are complementary to and synergistic with our business.
In addition, we expect to leverage the BioReference business and infrastructure to drive rapid and widespread uptake of our diagnostic products. Our strategy with respect to BioReference Laboratories is comprised of three pillars: the core business, digital health and strategic ventures. Because of the COVID-19 outbreak, we have added a fourth pillar as the COVID-19 pandemic impacted all parts of our organization. In support of this strategy:
•We have made significant investments to rebuild and reconfigure our main laboratory in Elmwood Park, NJ and have also made significant investments in our labs in Melbourne, Florida, Houston, Texas, California and the GeneDx laboratory in Maryland.
•We have facilitated increased patient access through preferred relationships with payors. We are part of the United Healthcare preferred lab network with access to approximately 45 million patients. We now also have access to Blue Cross/Blue Shield of Texas with another approximately 5.9 million patients and access to Blue Cross/Blue Shield of Alabama with another approximately 2 million patients.
•We intend to continue to expand large-scale COVID-19 screening programs nationwide. We offer customized solutions to different types of environments and to different types of clients. We are one of the largest providers of these programs, which include both PCR COVID-19 testing and point of care testing, across a variety of touch points, including the travel and leisure industry, airlines, cruise industry and education. BioReference has provided COVID-19 testing to the NFL, NBA, NHL, US National Soccer Teams, Winter X Games and the US Golf Association, among other organizations.
•We intend to continue to expand our offerings among our core laboratory service businesses in clinical, genetics, women’s health, oncology and urology.
•We intend to initiate digital health advances that aim to increase flexibility and convenience for patients. We recently introduced an important initiative in digital health through the launch of Scarlet™ Health, a fully-integrated digital platform providing mobile phlebotomy and laboratory services to patient’s homes, offices and other preferred locations.
•We intend to continue to expand and seek new strategic ventures to provide laboratory services for large health care groups and systems.
CORPORATE INFORMATION
We were originally incorporated in Delaware in October 1991 under the name Cytoclonal Pharmaceutics, Inc., which was later changed to eXegenics, Inc. (“eXegenics”). On March 27, 2007, we were part of a three-way merger with Froptix Corporation and Acuity Pharmaceuticals, Inc., both research and development companies. On June 8, 2007, we changed our name to OPKO Health, Inc. Our shares are publicly traded on the NASDAQ Stock Market under the ticker “OPK” and on the Tel Aviv Stock Exchange under the ticker “OPK”. Our principal executive offices are located in leased office space in Miami, Florida.
We currently manage our operations in two reportable segments: diagnostics and pharmaceuticals. The pharmaceutical segment consists of the pharmaceutical operations we operate in Chile, Mexico, Ireland, Israel and Spain and our pharmaceutical research and development operations. The diagnostics segment primarily consists of the clinical laboratory operations of BioReference, as well as our point-of-care operations. There are no significant inter-segment sales. We evaluate the performance of each segment based on operating profit or loss. There is no inter-segment allocation of interest expense and income taxes. Refer to Note 18 of our audited consolidated financial statements contained in this Annual Report on Form 10-K for financial information about our segments and geographic areas.
7
CURRENT PRODUCTS AND SERVICES AND RELATED MARKETS
Diagnostics
BioReference Laboratories
Through BioReference, one of the largest full service laboratories in the United States, we offer comprehensive laboratory testing services utilized by healthcare providers in the detection, diagnosis, evaluation, monitoring, and treatment of diseases, including esoteric testing, molecular diagnostics, anatomical pathology, genetics, women’s health and correctional healthcare. We market and sell these services to physician offices, clinics, hospitals, employers and governmental units nationally, with the largest concentration of business in the larger metropolitan areas in New York, New Jersey, Florida, Texas, Maryland, California, Pennsylvania, Delaware, Washington DC, Illinois and Massachusetts. BioReference is in network for over 80% of all U.S. insured lives.
BioReference has an almost 250-person sales and marketing team and operates a network of 128 active patient service centers.
Our BioReference laboratory testing business consists of routine testing and esoteric testing. Routine tests measure various health parameters, such as the functions of the heart, kidney, liver, thyroid and other organs, including such tests as blood cell counts, cholesterol levels, pregnancy, substance abuse and urinalysis. We typically operate 24 hours per day, 365 days per year and perform and report most routine test results within 24 hours.
The esoteric tests we perform require sophisticated equipment and materials, highly skilled personnel and professional attention. Esoteric tests are ordered less frequently than routine tests and typically are priced higher than routine tests. Esoteric tests include tests related to endocrinology, genetics and genomics, immunology, microbiology, HIV tests, molecular diagnostics, next generation sequencing, oncology, serology, and toxicology.
Through BioReference, we operate in the following highly specialized laboratory divisions:
•BioReference Laboratories. BioReference constitutes our core clinical testing laboratory offering automated, high volume routine testing services, COVID-19 testing, STAT testing, informatics, HIV, Hep C and other molecular tests.
•GenPath (Oncology). National oncology presence with expertise in cancer pathology and diagnostics, as well as molecular diagnostics. Core tests include FLOW, IHC, MicroArray, FISH, ISH, Morphology, and full service oncology.
•GenPath (Women’s Health). Innovative technology platform for sexually transmitted infections has enabled expansion nationally with specimens coming from 41 states, including Image Directed Paps analysis, HPV Plus, and STI Testing.
•GeneDx. Industry leading national laboratory for testing rare and ultra-rare genetic diseases with international reach, performing testing on specimens from more than 50 countries. We recently entered into an Agreement and Plan of Merger and Reorganization (the “GeneDx Merger Agreement”) with Sema4 Holdings Corp., a Delaware corporation (“Sema4”), pursuant to which Sema4 has agreed to acquire our wholly-owned subsidiary, GeneDx, Inc. (“GeneDx”), subject to the satisfaction of customary closing conditions.
We have one of the largest marketing staffs of any laboratory in the country with sales and marketing groups dedicated to urology, oncology, women’s health, genetic testing and correctional health, as well as cross-over groups selling to large institutions. Most of our sales personnel operate in a dual capacity, as sales and client support representatives, which we believe provides better customer service and a strong connection with our customers.
We are among the largest providers of large-scale COVID-19 screening programs across the country, with the capacity to run approximately 100,000 PCR tests a day. These large-scale screening programs include both PCR COVID-19 testing and point of care testing. We offer testing across a variety of touch points, including the travel and leisure industry, airlines, cruise industry and education. We have substantial relationships with local and state governments to provide testing services across all 50 states, with substantial service relationships in New York, New Jersey, and Michigan. Through our relationships with CVS and RiteAid, we provide access to testing services for the general public and currently provide testing at more than 700 CVS and RiteAid storefronts. BioReference has provided COVID-19 testing for the NFL, NBA, NHL, U.S. National Soccer Teams, Winter X Games and the US Golf Association, among other organizations.
8
We expect the clinical laboratory testing industry will continue to experience growth in testing volumes due to aging of the population in the U.S., patient awareness of the value of laboratory tests, a decrease in the cost of tests, the development of sophisticated and specialized tests for detection and management of disease, increased recognition of early detection and prevention as a means of reducing healthcare costs, and ongoing research and development in genetics and genomics and personalized medicine. Our mission is to be recognized by our clients as the premier provider of clinical laboratory testing, information and related services.
BioReference provides us with a significant diagnostics commercial infrastructure for marketing and sales that reached approximately 21 million and 19 million patients, respectively, in 2021 and 2020. In addition, its large team of managed care experts complement our efforts to ensure that payors recognize the value of our diagnostic and laboratory tests for reimbursement purposes. We continue to leverage the national marketing, sales and distribution resources of BioReference, along with its almost 250-person sales and marketing team, to enhance sales of and reimbursement for our 4Kscore test, a laboratory developed blood test that provides a personalized risk score for aggressive prostate cancer. We plan to leverage the BioReference commercial infrastructure and capabilities, as well as its extensive relationships with payors, to commercialize OPKO’s other diagnostic products under development.
4Kscore Test
We offer the 4Kscore test through BioReference. We began selling the 4Kscore test in the U.S. in March 2014 and in Europe and Mexico in September 2014 and January 2015, respectively. The 4Kscore test was approved by the FDA in December 2021 for use in men age 45 and older who have not had a prior prostate biopsy or a biopsy negative and have an age specific abnormal total PSA or abnormal digital rectal exam (DRE). The 4Kscore test is a laboratory developed test that measures the blood serum or plasma levels of four different prostate-derived kallikrein proteins: Total PSA, Free PSA, Intact PSA and Human Kallikrein-2 (“hK2”). These biomarkers are then combined with a patient’s age, optional DRE status (nodule / no nodule), and prior negative biopsy status (yes, prior negative biopsy / no prior biopsy) using a proprietary algorithm to calculate the risk (probability) of finding a Gleason Score 7 or higher prostate cancer. The four kallikrein panel of biomarkers utilized in the 4Kscore test is based on decades of research conducted by scientists at Memorial Sloan-Kettering Cancer Center and leading European institutions. Investigators at the Lund University, Sweden, University of Turku, Finland and Memorial Sloan Kettering Cancer Center, New York, have also demonstrated that the 4Kscore test can risk stratify the 20-year risk for development of prostate metastases and mortality in men who present at age 50 to 60 years old with an elevated PSA.
The 4Kscore test was developed by OPKO and validated in two prospective, blinded studies of 1,012 and 366 men, respectively. The first study was done in collaboration with 26 urology centers across the U.S. and the second study was conducted at eight VA centers in the U.S. with a predominantly African American cohort. African Americans are 1.7 times more likely to be diagnosed with prostate cancer than Caucasian men and 2.2 times more likely to die from the disease. Results showed that the 4Kscore test was highly accurate for predicting the presence of high-grade cancer (Gleason Score 7 or higher) prior to prostate biopsy, regardless of race. The full data from the blinded, prospective U.S. clinical validation studies have been published in peer reviewed medical journals.
The clinical data from both studies demonstrated the ability of the 4Kscore test to discriminate between men with high-grade, aggressive prostate cancer and those men who had no findings of cancer or had low-grade or indolent form of the disease. In separate clinical studies, use of the 4Kscore test led to 64.6% fewer biopsies and was able to discriminate between men with high-grade aggressive prostate cancer and those with no findings of cancer.
The National Comprehensive Cancer Network (“NCCN”) has included the 4Kscore test as a recommended test in its Guidelines for Prostate Cancer Early Detection since 2015. The panel making this recommendation concluded that the 4Kscore test is indicated for use prior to a first prostate biopsy, or after a negative biopsy, to assist patients and physicians in further defining the probability of high-grade cancer. In addition, the European Association of Urology (“EAU”) Prostate Cancer Guidelines Panel included the 4Kscore test in their Guidelines for Prostate Cancer since 2018, concluding that the 4Kscore, as a blood test with greater specificity over the PSA test, is indicated for use prior to a first prostate biopsy or after a negative biopsy to assist patients and physicians in further defining the probability of high-grade cancer.
The 4Kscore test has been granted a Category I CPT® code by the AMA (CPT Code 81539). A CPT code is used by insurance companies and government payors to describe health care services and procedures. A Category I CPT code is critical to facilitate reimbursement in government programs such as Medicare and Medicaid, as well as private insurance programs. Effective December 30, 2019, Novitas Solutions (“Novitas”), the local Medicare Administrative Contractor (“MAC”) for the 4Kscore testing laboratory in New Jersey, provided positive coverage through a local coverage determination (an “LCD”) with defined coverage criteria. Since that date, 4Kscore test orders meeting the coverage criteria have been reimbursed by Novitas and Medicare Advantage Health Plans.
9
Point-of-Care Diagnostics
OPKO Diagnostics, LLC (“OPKO Diagnostics”), formerly Claros Diagnostics, Inc., has developed a novel diagnostic instrument system to provide rapid, high performance blood test results in the point-of-care setting (the “Claros Analyzer”). The technology requires only a finger stick drop of blood introduced into the test cassette that can then be used to run a quantitative test. The instrument performs the tests on a disposable, one time usable cassette that is a microfluidics-based diagnostic test system. The credit card-sized test cassette works with a sophisticated desktop analyzer to provide high performance quantitative blood test results within 10-15 minutes and permits the transition of immunoassays from the centralized reference laboratory to the physician’s office, hospital nurses station, or other decentralized location.
We completed multiple in vitro analytical validation and field use tests for the PSA test in mid-2017 and filed the premarket approval application (“PMA”) for the Claros Analyzer and Sangia Total PSA Test with the FDA in November 2017. The FDA approved the PMA for the Sangia Total PSA Test using the Claros Analyzer in January 2019. The key clinical study with patients suspected of having prostate cancer found that the sensitivity of the Sangia Total PSA Test together with a DRE was improved versus the sensitivity of a DRE alone, to 91%, detecting 2.9 times the prostate cancers compared to a DRE alone. The FDA has categorized Sangia total PSA test as a moderately complex device under Clinical Laboratory Improvement Amendments of 1988 (“CLIA”).
We are currently evaluating commercialization strategies for the Claros system, and have several tests in development on the Claros system biomarkers.
Pharmaceutical Business
We currently have one commercial stage pharmaceutical product and several pharmaceutical compounds and technologies in various stages of research and development for a broad range of indications and conditions, including the following:
Renal Products-Rayaldee
Rayaldee is a patented extended release product containing 30 mcg of a prohormone, called calcifediol (25-hydroxyvitamin D3), for oral administration. We launched Rayaldee, our lead renal product, in the U.S. market in November 2016, following receipt in June 2016 of FDA approval for the treatment of SHPT in adults with stage 3 or 4 CKD and vitamin D insufficiency, defined as serum total 25-hydroxyvitamin D levels less than 30 ng/mL. The FDA approval of Rayaldee was supported by successful results from two identical randomized, double-blind, placebo-controlled, multi-site phase 3 studies which established the safety and efficacy of Rayaldee as a new treatment for SHPT in adults with stage 3 or 4 CKD and vitamin D insufficiency.
Vitamin D insufficiency arises in CKD due to the abnormal upregulation of CYP24A1, an enzyme that destroys vitamin D and its metabolites, from obesity and from many other causes as well. Studies in CKD patients have demonstrated that currently available over-the-counter and prescription vitamin D supplements cannot reliably and sufficiently raise blood vitamin D prohormone levels to effectively treat SHPT, a condition commonly associated with CKD in which the parathyroid glands secrete excessive amounts of PTH. Prolonged elevation of blood PTH causes excessive calcium and phosphorus to be released from bone, leading to elevated serum calcium and phosphorus levels, softening of the bones (osteomalacia) and calcification of vascular and renal tissues. SHPT affects 33% and 54% of patients with stage 3 and 4 CKD respectively, and approximately 95% of patients with stage 5 CKD.
We have a 57-person highly specialized sales, marketing and market access team dedicated to the commercialization of Rayaldee as of December 31, 2021. In the fourth quarter of 2021, total Rayaldee prescriptions decreased approximately 28.8% and 1.4% as compared to the fourth quarter of 2020 and the third quarter of 2021, respectively. Sales of Rayaldee have not increased in accordance with its expected growth trajectory as a result of challenges in onboarding new patients due to the COVID-19 pandemic. Efforts are underway to obtain broader commercial and Part D insurance coverage for Rayaldee. We have already achieved commercial and Medicare Part D formulary coverage for more than 79.9% of U.S. covered lives as of the end of 2021.
In May 2016, we entered into a development and license agreement (the “VFMCRP Agreement”) with Vifor Fresenius Medical Care Renal Pharma (“VFMCRP”) for the development and commercialization of Rayaldee in Europe, Canada, Mexico, Australia, South Korea and certain other international markets for the treatment of SHPT in patients with stage 3, 4 or 5 CKD and vitamin D insufficiency. The VFMCRP Agreement was later amended to exclude South Korea, the Middle East and all of the countries of Africa from the VFMCRP Territory (as defined in the VFMCRP Agreement), and further amended to include Japan as part of the VFMCRP Territory.
VFMCRP initiated the commercial launch of Rayaldee in Germany in February 2022 and has received marketing authorization from eleven European countries to date and is preparing for product launches in additional territories in 2022. The launch in Germany triggered a $3 million payment to EirGen Pharma Ltd. (“EirGen”), our wholly owned subsidiary.
10
In connection with the VFMCRP Agreement, the parties entered into a letter agreement pursuant to which EirGen granted to VFMCRP an exclusive option (the “Option”) to acquire an exclusive license under certain EirGen patents and technology to use, import, offer for sale, sell, distribute and commercialize the Product in the U.S. solely for the treatment of SHPT in dialysis patients with CKD and vitamin D insufficiency (the “Dialysis Indication”). Upon exercise of the Option, VFMCRP will reimburse EirGen for all of the development costs incurred by EirGen with respect to the Product for the Dialysis Indication in the U.S. VFMCRP would also pay EirGen up to an additional aggregate amount of $555 million of sales-based milestones upon the achievement of certain milestones and would be obligated to pay royalties at percentage rates that range from the mid-teens to the mid-twenties on sales of the Product in the U.S. for the Dialysis Indication. To date, VFMCRP has not exercised its option.
On June 18, 2021, EirGen and NICOYA Macau Limited (“Nicoya”), a Macau corporation and an affiliate of NICOYA Therapeutics, entered into a Development and License Agreement (the “Nicoya Agreement”) granting Nicoya the exclusive rights for the development and commercialization of extended release calcifediol (the “Nicoya Product”) in Greater China, which includes mainland China, Hong Kong, Macau, and Taiwan (collectively, the “Nicoya Territory”). Extended release calcifediol is marketed in the U.S. by OPKO under the tradename Rayaldee. The license grant to Nicoya covers the therapeutic and preventative use of the Nicoya Product for SHPT in non-dialysis and hemodialysis chronic kidney disease patients (the “Nicoya Field”).
OPKO and VFMCRP collaborated to complete a phase 2 study evaluating a higher strength dosage form of Rayaldee for the treatment of SHPT in hemodialysis patients. The study commenced in the 3rd quarter of 2018 and topline data were presented in an abstract titled “Initial Evaluation of High-Dose Extended-Release Calcifediol (ERC) in Patients with Stage 5 Chronic Kidney Disease on Hemodialysis” at the American Society of Nephrology’s Kidney Week Annual Meeting in November 2021.
In October 2020, we commenced a placebo controlled Phase 2 trial with Rayaldee as a treatment for mild-to-moderate COVID-19. In August 2021, the trial completed enrollment of 171 symptomatic patients from multiple U.S. sites and randomized in a 1:1 ratio for 4 weeks of treatment with Rayaldee or placebo and a 2-week follow-up. The trial ended in November 2021 and topline data were reported in December 2021 which indicated that improving vitamin D status with oral Rayaldee results in earlier resolution of respiratory symptoms associated with COVID-19. Data from the trial and potential next steps in the development program for this indication will be reviewed with the FDA later this year.
We have also completed a Phase 4 clinical trial comparing Rayaldee with three common treatment regimens for SHPT in adults with stage 3 or stage 4 CKD and vitamin D insufficiency. Preliminary results indicate that a daily dose of 60 micrograms of Rayaldee is the only one of four treatment regimens that reliably raises serum total 25-hydroxyvitamin D to the range of 50-100 mg/ml level required to effectively suppress elevated plasma parathyroid hormone levels in CKD patients. We presented interim results in an abstract titled “Comparison of Extended-Release Calcifediol (ERC), Immediate-Release Calcifediol, Cholecalciferol, and Paricalcitol for Treating Secondary Hyperparathyroidism (SHPT) in CKD” at the American Society of Nephrology’s Kidney Week Annual Meeting in October 2020.
We believe the CKD patient population is large and growing as a result of obesity, hypertension and diabetes; therefore this patient population represents a significant global market opportunity. According to the U.S. Renal Data System, CKD afflicts over 37 million people in the U.S., including more than 18 million patients with stage 3 or 4 CKD. In stage 5 CKD, kidney function is minimal to absent and most patients require regular dialysis or a kidney transplant for survival. An estimated 71-97% of CKD patients have vitamin D insufficiency which can lead to SHPT and its debilitating consequences. CKD continues to be associated with poor outcomes, reflecting the inadequacies of the current standard of care. We intend to develop and commercialize Rayaldee to constitute part of the foundation for a new and markedly improved standard of care for CKD patients having SHPT and/or hyperphosphatemia.
SARM
Through the acquisition of Transition Therapeutics, a Toronto-based biotechnology company (“Transition”), we acquired OPK88004, an orally administered selective androgen receptor modulator (“SARM”). The selective and antagonistic properties of OPK88004 on the prostate appear to be well suited to potentially reduce prostate hyperplasia and volume, as well as provide anabolic therapeutic benefits such as increased lean body mass and physical function, and decreased fat mass in specific patient populations. We believe that SARMs hold considerable promise as a new class of anabolic therapies for a variety of clinical indications, such as frailty and functional limitations associated with aging and chronic illnesses, cancer and osteoporosis.
Oxyntomodulin
11
Our internal product development program is also currently focused on developing a once weekly administered oxyntomodulin for type 2 diabetes and obesity. Our most advanced oxyntomodulin product candidate, OPK88003, a once-weekly administered peptide for the treatment of type 2 diabetes and associated obesity, is a dual agonist of the Glucagon-Like Peptide-1 (GLP-1) and glucagon receptors. The receptors play an integral role in regulating appetite, food intake, satiety and energy utilization in the body. Stimulating both of the receptors, OPK88003 has the potential to regulate blood glucose.
OPK88003 has been evaluated in a phase 2 study enrolling 420 type 2 diabetes subjects in a 24 week study consisting of a 12-week randomized blinded stage followed by a 12-week open-label stage. The study included four once-weekly dose arms of OPK88003 (10mg, 15mg, 30mg, 50mg), a placebo arm, and an active comparator arm (exenatide extended release – 2mg). The study was completed in February 2016.
Subjects receiving the highest dose of OPK88003 peptide once weekly in the study demonstrated significantly superior weight loss compared with currently approved extended release exenatide and placebo after 12 and 24 weeks of treatment. OPK88003 also provided a reduction in HbA1c, a marker of sugar metabolism, similar to exenatide at weeks 12 and 24.
We have evaluated OPK88003 in a dose escalation phase 2b trial in 110 type 2 diabetics where patients have been treated with a dose escalation regimen over 3 months intended to optimize dose levels, and increase body weight loss and reduce the adverse event profile, such as nausea and vomiting. The patients were treated for a total of 30 weeks in the study. In March 2019, we announced positive topline results from that phase 2b trial, which demonstrated that OPK88003 met the primary objective with a statistically significant lowering of hemoglobin A1c (HbA1c) after 30 weeks of treatment versus placebo as well as an important secondary endpoint, statistically significant weight loss versus placebo. The safety profile was similar to that expected for the incretin class of drugs, with GI side effects such as nausea, vomiting and diarrhea mostly mild and occurring during the dose-escalation phase.
On September 14, 2021, we and LeaderMed Health Group Limited (“LeaderMed”), a pharmaceutical development company with operations based in Asia, announced the formation of a joint venture under which we granted the joint venture exclusive rights to develop, manufacture and commercialize (a) OPK88003, an oxyntomodulin analog being developed for the treatment of obesity and diabetes, and (b) Factor VIIa-CTP, a novel long-acting coagulation factor being developed to treat hemophilia, in exchange for a 47% ownership interest in the joint venture. In addition, we have received an upfront payment of $1 million and will be reimbursed for clinical trial material and technical support it provides the joint venture.
LeaderMed will be responsible for funding the joint venture’s operations, development and commercialization efforts and will, with its syndicate partners, initially invest $11 million in exchange for a 53% ownership interest in the joint venture. We retain full rights to oxyntomodulin and Factor VIIa-CTP in all other geographies.
We believe oxyntomodulin has potential to be a safe, long term therapy for obesity and diabetes type II patients, representing significant market opportunities. More than 380 million are living with diabetes worldwide, of which approximately 90% have type II diabetes. According to the World Health Organization, there are more than 500 million severely overweight or obese people. In addition to diabetes and obesity, we are also considering development of this product candidate for additional indications, including treatment of non-alcoholic fatty liver disease and non-alcoholic steatohepatits.
Biologics-General
Our biologics business focuses on developing and commercializing longer-acting proprietary versions of already approved therapeutic proteins. One of our innovative platform technologies uses a short, naturally-occurring amino acid sequence, carboxl terminal peptide (“CTP”) which has the effect of slowing the removal from the body of the therapeutic protein to which it is attached. This CTP can be readily attached to a wide array of existing therapeutic proteins, stabilizing the therapeutic protein in the bloodstream and extending its life span without additional toxicity or loss of desired biological activity. We are using the CTP technology to develop new, proprietary versions of certain existing therapeutic proteins that have longer life spans than therapeutic proteins without CTP. We believe that our products will have greatly improved therapeutic profiles and distinct market advantages.
Somatrogon (hGH-CTP)
Our lead product candidate utilizing CTP, Somatrogon (hGH-CTP), is a recombinant human growth hormone product under development for the treatment of growth hormone deficiency (“GHD”), which is a pituitary disorder resulting in short stature in children and other physical ailments in both children and adults.
In December 2014, we entered into an exclusive worldwide agreement with Pfizer (the “Pfizer Transaction”) for the development and commercialization of hGH-CTP for the treatment of GHD in adults (“Adult GHD”) and in children (“Pediatric GHD”), as well as for the treatment of growth failure in children born small for gestational age (“SGA”). In connection with the Pfizer Transaction, we granted Pfizer an exclusive license to commercialize hGH-CTP worldwide, and we
12
received non-refundable and non-creditable upfront payments aggregating $295 million and are eligible to receive up to an additional $275 million upon the achievement of certain regulatory milestones. Upon the launch of hGH-CTP we are entitled to either regional, tiered gross profit sharing for both hGH-CTP and Pfizer’s Genotropin® once certain necessary pricing approvals are obtained, or tiered royalty payments on sales of Somatrogon with percentage rates ranging from the high teens to mid-twenties until such necessary pricing approval are obtained.
GHD occurs when the production of growth hormone, secreted by the pituitary gland, is disrupted. Since growth hormone plays a critical role in stimulating body growth and development, and is involved in the production of muscle protein and in the breakdown of fats, a decrease in the hormone affects numerous body processes. hGH is used for the long-term treatment of children and adults with inadequate secretion of endogenous growth hormone. The primary indications it treats in children are GHD, SGA, kidney disease, Prader-Willi Syndrome and Turner’s Syndrome. In adults, the primary indications are replacement of endogenous growth hormone and the treatment of AIDS-induced weight loss. Patients using hGH receive daily injections six or seven times a week. This is particularly burdensome for pediatric patients. We believe a significant market opportunity exists for a longer-lasting version of hGH that would require fewer injections.
Our phase 3 trial of hGH-CTP in pediatric patients was initiated in December 2016 and was completed in August 2019. The global study was a 224-patient study in Pediatric GHD patients designed to evaluate weekly treatment with hGH-CTP versus daily injections of Genotropin. hGH-CTP is delivered in a pen device in this multi-regional study in over 21 countries. The GHD subjects were treated weekly for 12 months. On October 21, 2019, we and Pfizer announced that the global phase 3 trial met its primary endpoint of non-inferiority to daily Genotropin® (somatropin) for injection, as measured by annual height velocity (“HV”) at 12 months. Results from this study demonstrated that treatment with hGH-CTP dosed once-weekly in pre-pubertal children with GHD was non-inferior to Genotropin® (somatropin) dosed once-daily with respect to HV at 12 months of treatment (the primary endpoint); the least square mean was higher in the hGH-CTP group (10.12 cm/year) than in the Genotropin® (somatropin) group (9.78 cm/year); the treatment difference (hGH-CTP—Genotropin® (somatropin)) in HV (cm/year) was 0.33 with a two-sided 95% confidence interval of the difference of (-0.39, 1.05). In addition, change in height standard deviation scores at six and 12 months, key secondary endpoints, were higher in the hGH-CTP dosed once-weekly cohort in comparison to the Genotropin® (somatropin) dosed once-daily cohort. hGH-CTP was generally well tolerated in this study and comparable to that of Genotropin® (somatropin) dosed once-daily with respect to the types, numbers and severity of the adverse events observed between the treatment arms.
We believe hGH-CTP represents a significant advancement in the treatment of children with GHD compared to the current standard of one injection per day that could enhance a patient’s adherence to treatment and quality of life.
In addition to the phase 3 pediatric study, we have continued without interruption our ongoing phase 2 pediatric open label extension study for hGH-CTP. Most of the phase 2 pediatric patients have been treated with hGH-CTP for more than six years, and some patients for more than seven years. We have switched all of the pediatric patients in this study to the disposable pen device. A 44-patient Phase 3 study in Pediatric GHD patients in Japan was completed in the first quarter of 2020. The Japan Phase 3 clinical trial met its primary and secondary objectives, and demonstrated that the efficacy and safety of hGH-CTP administered weekly was comparable to Genotropin® as measured by annual height velocity after 12 months of treatment in pre-pubertal children with GHD. The findings were consistent with the results previously reported in the Phase 3 global study. The least squared means for the annual height velocity was higher in the Somatrogon group (9.65 cm/year) than in the Genotropin group (7.87 cm/year).
We submitted the initial Biologics License Application (“BLA”) with the FDA for approval of Somatrogon in the United States and Pfizer recently received a Complete Response Letter. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine an appropriate path forward. Regulatory applications for Somatrogon have been submitted to several countries around the world for review. In February 2022, the European Commission granted marketing authorization in the European Union for Somatrogon under the brand name NGENLA® to treat children and adolescents from as young as 3 years of age with growth disturbance due to insufficient secretion of growth hormone. In January 2022, the Ministry of Health, Labour and Welfare in Japan approved NGENLA® (Somatrogon) for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. In October 2021, Health Canada approved NGENLA® for the long-term treatment of pediatric patients who have growth hormone deficiency, and in November 2021, Australia’s Therapeutic Goods Administration approved NGENLA® for the long-term treatment of pediatric patients with growth disturbance due to insufficient secretion of growth hormone.
13
In December 2016, we announced preliminary topline data from our phase 3, double blind, placebo controlled study of hGH-CTP in adults with GHD. The multinational, multi-center study, which utilized a 2:1 randomization between hGH-CTP and placebo, enrolled 203 subjects, 198 of whom received at least one dose of study treatment. Treatment was administered through a weekly injection. The topline results showed:
•The active group had a mean change in trunk fat mass of -0.4kg and placebo group was 0;
•There was no statistically significant difference (≤ 0.05 (p value)) between the active and placebo group;
•97% of hGH-CTP vs 6% of placebo group showed IGF-1 normalization; and
•The safety profile of hGH-CTP is consistent with that observed with those treated with daily growth hormone.
Although there was no statistically significant difference between hGH-CTP and placebo on the primary endpoint of change in trunk fat mass from baseline to 26 weeks, after unblinding the study, we identified an exceptional value of trunk fat mass reduction in the placebo group that may have affected the primary outcome. We have completed post-hoc sensitivity analyses to evaluate the influence of outliers on the primary endpoint results using multiple statistical approaches. Analyses that excluded outliers showed a statistically significant difference between hGH-CTP and placebo on the change in trunk fat mass. Additional analyses that did not exclude outliers showed mixed results. Further, significant changes were observed with hGH-CTP treatment in secondary endpoints such as lean body mass and compared to placebo. We believe there is a path for submission of a BLA with respect to hGH-CTP for adults with GHD on the basis that the FDA may assess the totality of the data, including all relevant efficacy and safety data in adult and pediatric patients. We plan to continue to assess with Pfizer the regulatory strategy for the adult indication going forward, including the timing of a possible submission.
Factor VIIa-CTP
In addition to hGH-CTP, we have a product candidate to extend the duration of the biological activity of Factor VIIa (hemophilia) using our CTP technology. In February 2013, the FDA granted orphan drug designation to our longer-acting version of clotting Factor VIIa, Factor VIIa-CTP, for the treatment of bleeding episodes in patients with hemophilia A or B with inhibitors to Factor VIII or Factor IX. We have completed a phase 1 single dose subcutaneously administered Factor VIIa-CTP study in healthy volunteers and a phase 2a single dose trial in Hemophilia A patients. Factor VIIa-CTP exhibited a positive safety profile in both hemophiliac patients and healthy subjects following a single IV or subcutaneous injection respectively. Pharmacodynamic assessment of coagulation markers demonstrated pharmacological activity of Factor VIIa-CTP with an extended response. We will need to conduct additional toxicity studies before we are in a position to present a clinical study plan.
We also entered into a joint venture with LeaderMed on September 14, 2021, under which we granted the joint venture exclusive rights to develop, manufacture and commercialize (a) OPK88003, an oxyntomodulin analog being developed for the treatment of obesity and diabetes, and (b) Factor VIIa-CTP, a novel long-acting coagulation factor being developed to treat hemophilia.
Early Stage Biologics Pipeline
In addition to hGH-CTP and Factor VIIa-CTP, we believe that the CTP technology may also be broadly applicable to other therapeutic proteins in the market and provide a reduction in the number of injections required for treatment. We are currently engaged in research and development efforts to use the CTP technology for development of a long-acting CTP-IGF-1 for the Treatment of Severe Primary IGF-1 Deficiency.
In addition to development efforts using the CTP platform, we are also focused on broadening the approaches used to develop long acting therapies for once weekly therapies in rare diseases.
APIs
FineTech Pharmaceutical, Ltd. (“FineTech”), is our Israeli-based subsidiary that develops and produces high value, high potency specialty APIs. FineTech currently manufactures commercial APIs for sale or license to pharmaceutical companies in Latin America, Canada, Europe and Israel. We believe that FineTech’s significant know-how and experience with analytical chemistry and organic syntheses, together with its production capabilities, may play a valuable role in the development of our pipeline of proprietary molecules and compounds for diagnostic and therapeutic products, while providing revenues and profits from its existing API business.
14
Oligonucleotide Therapeutics
OPKO CURNA’s platform technology utilizes a short, single strand oligonucleotide to increase production of endogenous protein through interference with non-coding RNA’s or natural antisense. This strategy contrasts with established approaches which down-regulate protein production. CURNA has designed a novel type of therapeutic modality, termed AntagoNAT, and has initially demonstrated this approach for up-regulation of several therapeutically relevant proteins in in vitro and animal models.
We have filed an investigational new drug application, or IND, for a lead compound to treat Dravet Syndrome. Further preclinical work has been requested by the FDA prior to initiation of a first clinical study. Orphan disease designations have been granted by FDA and EMA.
On July 6, 2021, we entered into an exclusive license agreement (the “CAMP4 Agreement”) with CAMP4 Therapeutics Corporation (“CAMP4”), pursuant to which we granted to CAMP4 an exclusive license to develop, manufacture, commercialize or improve therapeutics utilizing the AntagoNAT technology, which includes the molecule for the treatment of Dravet syndrome, together with any derivative or modification thereof (the “CAMP4 Licensed Compound”) and any pharmaceutical product that comprises or contains the CAMP4 Licensed Compound, alone or in combination with one or more other active ingredients (“CAMP4 Licensed Product”), worldwide. The CAMP4 Agreement grant covers human pharmaceutical, prophylactic, and therapeutic and certain diagnostic uses.
Commercial Operations
We may continue to leverage our global commercialization expertise to pursue acquisitions of commercial businesses that will both drive our growth and provide geographically diverse sales and distribution opportunities. During 2015, we acquired EirGen, a specialty pharmaceutical company based in Ireland. EirGen is focused on the development and commercial supply of high potency, high barrier to entry, pharmaceutical products. Through its facility in Waterford, Ireland, EirGen currently manufactures high potency pharmaceutical products and exports to over 50 countries. High potency drugs such as those used for cancer chemotherapy are typically unsuitable for manufacture in normal multi-product facilities due to cross contamination risks.
To date, EirGen and its commercial partners have filed several product applications with the FDA in Europe and in Japan. EirGen has a strong research and development portfolio of high barrier to entry drugs and we expect to expand its drug portfolio. We believe EirGen will play an important role in the development, manufacturing, distribution and approval of a wide variety of drugs in a variety of dosage forms with an emphasis on high potency products.
OPKO Health Europe (previously Farmadiet Group Holding, S.L.) operates primarily in Spain and has more than 20 years of experience in the development, manufacture, marketing and sale of pharmaceutical, nutraceutical and veterinary products in Europe.
OPKO Mexico (previously Pharmacos Exakta S.A. de C.V.), is engaged in the manufacture, marketing, sale and distribution of ophthalmic and other pharmaceutical products to private and public customers in Mexico. OPKO Mexico is commercializing food supplements and over the counter products, and manufactures and sells products primarily in the generics market in Mexico, although it also has some proprietary products as well.
OPKO Chile (previously Pharma Genexx, S.A.) markets, sells and distributes pharmaceutical products to the private, hospital, pharmacy and public institutional markets in Chile for a wide range of indications, including, cardiovascular products, vaccines, antibiotics, gastro- intestinal products and hormones, among others. ALS Distribuidora Limitada (“ALS”) is engaged in the business of importation, commercialization and distribution of pharmaceutical products for private markets in Chile. ALS started operations in 2009 as the exclusive product distributor of Arama Laboratorios y Compañía Limitada (“Arama”), a company with more than 20 years of experience in the pharmaceutical products market. In connection with the acquisition of ALS, OPKO acquired all of the product registrations and trademarks previously owned by Arama, as well as the Arama name. We distribute food supplements and over the counter products through Arama.
Strategic Investments
We have and may continue to make investments in other early stage companies that we perceive to have valuable proprietary technology and significant potential to create value for OPKO as a shareholder.
RESEARCH AND DEVELOPMENT EXPENSES
15
During the years ended December 31, 2021, 2020, and 2019, we incurred $76.9 million, $75.3 million, and $117.9 million, respectively, of research and development expenses related to our various product candidates. During the years ended December 31, 2021, 2020, and 2019, our research and development expenses primarily consisted of hGH-CTP and Rayaldee development programs.
INTELLECTUAL PROPERTY
We believe that technology innovation is driving breakthroughs in healthcare. We have adopted a comprehensive intellectual property strategy which blends the efforts to innovate in a focused manner with the efforts of our business development activities to strategically in-license intellectual property rights. We develop, protect, and defend our own intellectual property rights as dictated by the developing competitive environment. We value our intellectual property assets and believe we have benefited from early and insightful efforts at understanding diagnostics, as well as the disease and the molecular basis of potential pharmaceutical intervention.
We actively seek, when appropriate and available, protection for our products and proprietary information by means of U.S. and foreign patents, trademarks, trade secrets, copyrights, and contractual arrangements. Patent protection in the pharmaceutical and diagnostic fields, however, can involve complex legal and factual issues. There can be no assurance that any steps taken to protect such proprietary information will be effective.
We own or license-in thousands of U.S. and foreign patents and applications for our products, product candidates and our outlicensed product candidates. These patents cover pharmaceuticals, diagnostics and other products and their uses, pharmaceutical and diagnostic compositions and formulations and product manufacturing processes. Our patents are filed in various locations worldwide as is appropriate to the particular patent and its use.
Rayaldee
We have multiple U.S. patent families relating to Rayaldee. These patents are also filed in multiple countries worldwide. One patent family claims a sustained release oral dosage formulation and a method of treating 25-hydroxyvitamin D insufficiency or deficiency and will not expire until at least February 2027. A second patent family claims a method of administering 25-hydroxyvitamin D3 by controlled release, a formulation for controlled release of a vitamin D compound, a controlled release oral dosage formulation of a vitamin D compound and a method of treatment, and will not expire until at least April 2028. We also have additional patents and patent applications pending relating to the sustained release formulation and its use which will expire in 2034. The patents issued in the U.S. covering Rayaldee are listed in the Approved Drug Products with Therapeutic Equivalence Evaluations, or the Orange Book. OPKO and/or its affiliates have entered into exclusive license agreements with respect to Rayaldee patents in certain territories outside of North America with VFMCRP (Europe and many other countries throughout the rest of the world), and Nicoya Macau Limited (China). We intend to seek patent term extensions in those countries for which such protection is potentially available. We also continue to file and seek patent protection on various uses of extended release dosage forms of 25-hydroxyvitamin D3 and new formulations of this drug. EirGen’s patent publication US2021/0308151 disclosing and claiming the use of 25-hydroxyvitamin D3 and controlled release formulations thereof to treat SARS-CoV-2 infection was published on October 7, 2021.
Somatrogon (hGH-CTP)
The hGH-CTP line of patents, which is exclusively licensed to Pfizer, includes multiple U.S. patent families that cover modified human grown hormone (Somatrogon), uses of Somatrogon in adult and pediatric patient populations, and methods of making Somatrogon. Equivalent patents have also been filed in multiple countries around the world. One patent family covers certain CTP modified hGH polypeptides relating to growth hormones and their method of use and expires in February of 2027 (with the exception of two U.S. patents, namely US 8304386 and US 8097435, which expire in January 2028 and April 2027, respectively, due to Patent Term Adjustment for each). Additional U.S. patent applications are pending which cover Somatrogon formulations, methods of manufacture and pediatric dosing regimens and, if granted, would expire in 2033. Equivalent patents are granted in Europe and Japan and which expire in 2032 and 2034. A subset of cases in the patent estate covers cytokine-based polypeptides relating to human growth hormone treatment and will expire in February 2027 (in the U.S., these cases include registered patents 8,048,849; 8,426,166; 8,999,670; and 9,896,494, and no Patent Term Adjustment was issued). Multiple other U.S. patents cover Somatrogon and its uses or methods of making including U.S. Pat. Nos. 7,553,941; 8,450,269; 8,946,155; 10,351,615; and 11,197,915, where no Patent Term Adjustment was awarded by the USPTO. The equivalent foreign patents and applications are granted or pending in several major market countries and regions. In addition to the CTP patents and applications licensed to Pfizer, OPKO has multiple patent families covering similar biologicals with patents and applications pending in the U.S. and internationally. Patent term extensions will be sought in those countries where Somatrogon is approved.
16
OPK88003 and OPK88004
In 2016, we acquired Transition which is developing multiple drug candidates that include OPK88003 (a long acting oxyntomodulin) and OPK88004 (SARM), each of which are licensed from Eli Lilly and have granted patents worldwide covering the compounds and their use in their respective indications. U.S. Pat. No. 8367607 covers OPK88003 and expires in December 2030, without extension. OPKO has also filed a formulation patent on a long acting oxyntomodulin formulation. U.S. Pat. No. 7968587 covers OPK88004 (SARM) and expires, without extension, in November 2027. In addition to the molecule patent covering the selective androgen receptor modulator, Transition Therapeutics exclusively licensed a method of use patent family covering its use in treating androgen deprivation therapy associated symptoms. These patents expire in 2035. OPKO has also filed additional patent applications on expanded uses of OPK88004. In addition, Transition and its affiliates have patented compounds (scyllo-inositol) for the treatment of Alzheimer’s disease. The patents are pending or granted in many countries of the world. OPKO and/or its affiliates or licensees will seek all available patent term extensions for our product candidates and products.
Because the patent positions of pharmaceutical, biotechnology, and diagnostics companies are highly uncertain and involve complex legal and factual questions, the patents owned and licensed by us, or any future patents, may not prevent other companies from developing similar or therapeutically equivalent products or ensure that others will not be issued patents that may prevent the sale of our products or require licensing and the payment of significant fees or royalties. Furthermore, to the extent that any of our future products or methods are not patentable, that such products or methods infringe upon the patents of third parties, or that our patents or future patents fail to give us an exclusive position in the subject matter claimed by those patents, we will be adversely affected. We may be unable to avoid infringement of third party patents and may have to obtain a license, defend an infringement action, or challenge the validity of the patents in court. A license may be unavailable on terms and conditions acceptable to us, if at all. Patent litigation is costly and time consuming, and we may be unable to prevail in any such patent litigation or devote sufficient resources to even pursue such litigation.
LICENSES AND COLLABORATIVE RELATIONSHIPS
Our strategy is to develop a portfolio of product candidates through a combination of internal development, acquisition, and external partnerships. Collaborations are key to our strategy and we continue to build relationships and forge partnerships in various areas where unmet medical need and commercial opportunities exist. In October 2017, we entered into a license and development agreement with JT for the development and commercialization of Rayaldee in Japan for the treatment of SHPT in non-dialysis and dialysis patients with CKD. Immediately following JT’s termination of this Agreement in May 2021, VFMCRP entered into an agreement with OPKO pursuant to which VFMCRP assumed JT’s rights in Rayaldee in Japan. Under the VFMCRP Agreement, as amended from time to time, we have a license and collaboration agreement for the development and commercialization of Rayaldee in Europe, Canada, Australia, and certain other international markets for the treatment of SHPT in patients with CKD and vitamin D insufficiency. In June 2021, we entered into a license agreement with Nicoya to distribute and sell Rayaldee in China and certain other countries. In July 2021, we licensed out our AntagoNAT portfolio owned by CURNA, INC. to CAMP4. In September 2021, we also entered into specific arrangements with LeaderMed in certain countries in Asia with respect to OPK-88003 and Factor VIIa. In November 2021, EirGen licensed out Rayaldee patent estate to Progenetics Ltd. to distribute Rayaldee in Israel. In December 2014, we entered into the Pfizer Transaction for the development and commercialization of our long-acting hGH-CTP for the treatment of GHD in adults and children, as well as for the treatment of growth failure in children born small for gestational age. Previously, we (or entities we have acquired) have completed strategic licensing transactions with the President and Fellows of Harvard College, Academia Sinica, The Scripps Research Institute, TESARO, INEOS Healthcare, and Arctic Partners, among others.
COMPETITION
The pharmaceutical and diagnostic testing industries are highly competitive and require an ongoing, extensive search for technological innovation. The industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. They also require, among other things, the ability to effectively discover, develop, test and obtain regulatory approvals for products, as well as the ability to effectively commercialize, market and promote approved products.
Numerous companies, including major pharmaceutical companies, specialty pharmaceutical companies and specialized biotechnology companies, are engaged in the development, manufacture and marketing of pharmaceutical products competitive with those that we are or intend to commercialize ourselves and through our partners. Competitors to our diagnostics business include major diagnostic companies, reference laboratories, molecular diagnostic firms, universities and research institutions. Most of these companies have substantially greater financial and other resources, larger research and development staffs and more extensive marketing and manufacturing organizations than ours. This enables them, among other things, to make greater research and development investments and efficiently utilize their research and development costs, as well as their marketing and promotion costs, over a broader revenue base. This also provides our competitors with a competitive advantage in connection with the highly competitive product acquisition and product in-licensing process, which may include auctions in
17
which the highest bidder wins. Our competitors may also have more experience and expertise in obtaining marketing approvals from the FDA and other regulatory authorities. In addition to product development, testing, approval, and promotion, other competitive factors in the pharmaceutical and diagnostics industry include industry consolidation, product quality and price, product technology, reputation, customer service, and access to technical information.
With regard to our pharmaceutical products, Rayaldee’s competition includes, among other products, activated (1-alpha-hydroxylated) vitamin D analogs such as calcitriol, doxercalciferol, and paricalcitol, and vitamin D supplements such as ergocalciferol and cholecalciferol. Although we believe that Rayaldee offers substantial benefits over these products, Rayaldee may be competing with these and other lower priced products and products which are marketed by larger pharmaceutical companies with substantially greater resources.
We are aware of a number of pharmaceutical and biopharmaceutical companies that have commenced clinical studies of products or have successfully commercialized products addressing areas that we are targeting with our long acting hGH-CTP. For example, several companies are developing sustained release or long-acting products for the treatment of GHD, and a number of companies currently market generic daily human growth hormone products for GHD.
In our clinical laboratory operations, we compete with three types of providers in a highly fragmented and competitive industry: hospital laboratories, physician-office laboratories and other independent clinical laboratories. Our major competitors in the New York metropolitan area are two of the largest national laboratories, Quest Diagnostics and Laboratory Corporation of America. Although we are much smaller than these national laboratories, we believe that we compete successfully with them in our region due to our innovative testing services and our level of service. We believe our responses to medical consultation are faster and more personalized than those of the national laboratories. Our client service staff deals only with basic technical questions and those that have medical or scientific significance are referred directly to our senior scientists and medical staff.
We are commercializing our 4Kscore product in the U.S., Europe and Mexico in a laboratory setting and seek to capitalize on commercialization opportunities for our proprietary diagnostic point-of-care system by transitioning laboratory-based tests, including PSA and other tests to our point-of-care system. Competitors to our diagnostics business are many and include major diagnostic companies, molecular diagnostic firms, universities, and research institutions.
Pricing and reimbursement coverage positions could substantially impact the competitiveness of the 4Kscore test and our other diagnostic products. Our ability to commercialize our pharmaceutical and diagnostic test product candidates and compete effectively will depend, in large part, on:
•our ability to meet all necessary regulatory requirements to advance our product candidates through clinical trials and the regulatory approval process in the U.S. and abroad;
•the perception by physicians and other members of the health care community of the safety, efficacy, and benefits of our products compared to those of competing products or therapies;
•our ability to manufacture products we may develop on a commercial scale;
•the effectiveness of our sales and marketing efforts;
•the willingness of physicians to adopt a new diagnostic or treatment regimen represented by our technology;
•our ability to secure reimbursement for our product candidates;
•the price of the products we may develop and commercialize relative to competing products;
•our ability to accurately forecast and meet demand for our product candidates if regulatory approvals are achieved;
•our ability to develop a commercial scale infrastructure either on our own or with a collaborator, which would include expansion of existing facilities, including our manufacturing facilities, development of a sales and distribution network, and other operational and financial systems necessary to support our increased scale;
•our ability to maintain a proprietary position in our technologies; and
•our ability to rapidly expand the existing information technology infrastructure and configure existing operational, manufacturing, and financial systems (on our own or with third party collaborators) necessary to support our increased scale, which would include existing or additional facilities and or partners.
GOVERNMENT REGULATION
The U.S. government regulates healthcare through various agencies, including but not limited to the following: (i) the FDA, which administers the Federal Food, Drug and Cosmetic Act (“FDCA”), as well as other relevant laws; (ii) the Centers for Medicare & Medicaid Services (“CMS”), which administers the Medicare and Medicaid programs; (iii) the Office of
18
Inspector General (“OIG”), which enforces various laws aimed at curtailing fraudulent or abusive practices, including by way of example, the Anti-Kickback Statute, the Physician Self-Referral Law, commonly referred to as the Stark law, the Civil Monetary Penalty Law (including the beneficiary inducement prohibition) (“CMP”), and the laws that authorize the OIG to exclude healthcare providers and others from participating in federal healthcare programs; and (iv) the Office of Civil Rights, which administers the privacy aspects of the Health Insurance Portability and Accountability Act of 1996. All of the aforementioned are agencies within the Department of Health and Human Services (“HHS”). Healthcare is also provided or regulated, as the case may be, by the Department of Defense through its TRICARE program, the Department of Veterans Affairs, especially through the Veterans Health Care Act of 1992, the Public Health Service within HHS under Public Health Service Act § 340B (42 U.S.C. § 256b), the Department of Justice through the Federal False Claims Act and various criminal statutes, and state governments under the Medicaid and other state sponsored or funded programs and their internal laws regulating all healthcare activities.
The testing, manufacture, distribution, advertising, and marketing of drug and diagnostic products and medical devices, as well as the performance of clinical testing services, are subject to extensive regulation by federal, state, and local governmental authorities in the U.S., including the FDA, and by similar agencies in other countries. Any drug, diagnostic, or device product that we develop must receive all relevant regulatory approvals or clearances, as the case may be, before it may be marketed in a particular country.
Clinical Laboratory Operations
Our clinical laboratory operations are subject to regulations, which are designed to ensure the quality and reliability of clinical laboratories by mandating specific standards in the areas of personnel qualifications, administration and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. Laboratories must undergo on-site surveys at least every two years, which may be conducted by CMS under the CLIA program or by a private CMS approved accrediting agency. The sanction for failure to comply with CLIA requirements may be suspension, revocation or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as significant fines and/or criminal penalties. We are also subject to regulation of laboratory operations under state clinical laboratory laws. State clinical laboratory laws may require that laboratories and/or laboratory personnel meet certain qualifications, specify certain quality controls or require maintenance of certain records. Certain states, such as New York, California, Maryland, Pennsylvania, and Rhode Island, each require that we obtain licenses to test specimens from patients residing in those states and additional states may require similar licenses in the future. Only Washington and New York State are exempt under CLIA, as these states have established laboratory quality standards at least as stringent as CLIA’s. Potential sanctions for violation of these statutes and regulations include significant fines and the suspension or loss of various licenses, certificates and authorizations.
Our clinical laboratory operations are subject to complex laws, regulations and licensure requirements relating to billing and payment for laboratory services, sales and marketing interactions with ordering physicians and other health care providers, security and confidentiality of health information, and environmental and occupational safety, among others. Changes in regulations often increase the cost of testing or processing claims. Also, these laws may be interpreted or applied by a prosecutorial, regulatory or judicial authority in a manner that could require us to make changes in our operations, including in our pricing, billing and/or marketing practices in a manner that could adversely affect operations.
Drug Development
The regulatory process, which includes overseeing preclinical studies and clinical trials of each pharmaceutical compound to establish its safety and efficacy and confirmation by the FDA that good laboratory, clinical, and manufacturing practices were maintained during testing and manufacturing, can take many years, requires the expenditure of substantial resources, and gives larger companies with greater financial resources a competitive advantage over us. Delays or terminations of clinical trials that we undertake would likely impair our development of product candidates. Delays or terminations could result from a number of factors, including stringent enrollment criteria, slow rate of enrollment, size of patient population, having to compete with other clinical trials for eligible patients, geographical considerations, failure to meet anticipated clinical success, patient safety concerns, and others.
Although accelerated pathways for approval exist for certain drugs, generally, FDA review processes can be lengthy and unpredictable, and we may encounter delays or rejections of our applications when submitted. Generally, in order to gain FDA approval, we must first conduct preclinical studies in a laboratory and in animal models to obtain preliminary information on a compound and to identify any safety problems. The results of these studies are submitted as part of an IND application that the FDA must review before human clinical trials of an investigational drug can commence.
Clinical trials are normally done in three sequential phases and generally take two to five years or longer to complete. phase 1 consists of testing the drug product in a small number of humans, normally healthy volunteers, to determine preliminary safety and tolerable dose range. Phase 2 usually involves studies in a limited patient population to evaluate the
19
effectiveness of the drug product in humans having the disease or medical condition for which the product is indicated, determine dosage tolerance and optimal dosage, and identify possible common adverse effects and safety risks. Phase 3 consists of additional controlled testing at multiple clinical sites to establish clinical safety and effectiveness in an expanded patient population of geographically dispersed test sites to evaluate the overall benefit-risk relationship for administering the product and to provide an adequate basis for product labeling. Phase 4 clinical trials may be conducted- and are sometimes required - after approval to gain additional experience from the treatment of patients in the intended therapeutic indication. There are also certain situations when drugs and biologics are eligible for one of FDA’s expedited approval programs, designed to shorten review and development time.
After completion of clinical trials of a new drug product, FDA and foreign regulatory authority marketing approval must be obtained. Assuming that the clinical data support the product’s safety and effectiveness for its intended use, a BLA or an NDA is submitted to the FDA for its review. Since the early 1990s, the FDA has managed a user fee program whereby sponsors of drug applications pay a fee to the agency and the agency commits to meeting a series of performance goals designed to reduce drug review times. Generally, it takes one to three years to obtain approval. If questions arise during the FDA review process, approval may take a significantly longer period of time. The testing and approval processes require substantial time and effort and we may not receive approval on a timely basis, if at all, or the approval that we receive may be for a narrower indication than we had originally sought, potentially undermining the commercial viability of the product. Even if regulatory approvals are obtained, a marketed product is subject to continual review, and later discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product or withdrawal of the product from the market as well as possible civil or criminal sanctions. For marketing outside the U.S., we also will be subject to foreign regulatory requirements governing human clinical trials and marketing approval for pharmaceutical products. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary widely from country to country.
In addition to clinical trial rules, FDA imposes other requirements on applicants including obligations related to Good Manufacturing Practices (GMPs), proper labeling, and other issues related to manufacturing and marketing a drug.
Other than NGENLA (Somatrogon), which has been approved in the EU, Japan, Canada and Australia, Rayaldee is our only pharmaceutical product under development that has been approved for marketing in the U.S. or elsewhere. We may not be able to obtain regulatory approval for any of our other products under development in a timely manner, if at all. Failure to obtain requisite governmental approvals or failure to obtain approvals of the scope requested will delay or preclude us, or our licensees or marketing partners, from marketing our products, or limit the commercial use of our products, and thereby would have a material adverse effect on our business, financial condition, and results of operations. See “Risk Factors — The results of pre-clinical trials and previous clinical trials for our products may not be predictive of future results, and our current and planned clinical trials may not satisfy the requirements of the FDA or other non-U.S. regulatory authorities.”
Device Development
Medical devices are subject to varying levels of premarket regulatory control, the most comprehensive of which requires human clinical trials be conducted before a device receives approval for commercial distribution. The FDA classifies medical devices into one of three classes based upon their risk profile (both to the patient and provider): Class I devices are relatively simple “low risk” technologies, and can be manufactured and distributed with general controls without a premarket clearance or approval from the FDA; Class II devices are somewhat more complex “moderate risk” devices, and require greater scrutiny from the agency, requiring a premarket clearance from the FDA before market entry; Class III devices are “high risk” technologies inserted or implanted in the body, intended to treat life sustaining functions. These Class III technologies require a premarket approval from the FDA before market entry.
In the U.S., a company generally can obtain permission to distribute a new device in one of two ways. The first applies to a Class II device that is substantially equivalent to a device first marketed prior to May 1976, or to another device marketed after that date, but which was substantially equivalent to a pre-May 1976 device. To obtain FDA permission to distribute the device, a company generally must submit a section 510(k) premarket notification, and receive an FDA order finding substantial equivalence to a predicate device (pre-May 1976 or post-May 1976 device that was substantially equivalent to a pre-May 1976 device) and permitting commercial distribution of that device for its intended use. A 510(k) submission must provide information supporting a claim of substantial equivalence to the predicate device. If clinical data from human experience are required to support the 510(k) submission, these data must be gathered in compliance with investigational device exemption (“IDE”), regulations for investigations performed in the U.S. The 510(k) process is normally used for products of the type that the Company proposes distributing. The FDA review process for premarket notifications submitted pursuant to section 510(k) takes, on average, about 90 days, but it can take substantially longer if the FDA has concerns, and there is no guarantee that the FDA will “clear” the device for marketing, in which case the device cannot be distributed in the U.S. There is also no
20
guarantee that the FDA will deem the applicable device subject to the 510(k) process, as opposed to the more time-consuming, resource-intensive and problematic, PMA process described below.
The second, more comprehensive, PMA process, which can take a year or longer, applies to a new device that is not substantially equivalent to a pre-1976 product or that is to be used in supporting or sustaining life or preventing impairment. These devices are normally Class III devices. For example, most implantable devices are subject to the approval process. Two steps of FDA approval are generally required before a company can market a product in the U.S. that is subject to approval, as opposed to clearance. First, a company must comply with IDE regulations in connection with any human clinical investigation of the device. These regulations permit a company to undertake a clinical study of a “non-significant risk” device without formal FDA approval. Prior express FDA approval is required if the device is a significant risk device. Second, the FDA must review the company’s PMA application, which contains, among other things, clinical information acquired under the IDE. The FDA will approve the PMA application if it finds there is reasonable assurance that the device is safe and effective for its intended use. The PMA process takes substantially longer than the 510(k) process and it is conceivable that the FDA would not agree with our assessment that a device that we propose to distribute should be a Class I or Class II device. If that were to occur we would be required to undertake the more complex and costly PMA process. However, for either the 510(k) or the PMA process, the FDA could require us to run clinical trials, which would pose all of the same risks and uncertainties associated with the clinical trials of drugs, described above.
In December of 2016, Congress enacted the 21st Century Cures Act (P.L. 114-255) which contained provisions establishing a new Breakthrough Device pathway to allow faster patient access to devices and breakthrough technologies that provide for more effective treatment or diagnosis for life-threatening or irreversibly debilitating diseases, for which no approved or cleared treatment exists or that offer significant advantages over existing approved or cleared alternatives. FDA has just begun to implement this program and it is not clear if any of our products would be eligible.
Even when a clinical study has been approved by the FDA or deemed approved, the study is subject to factors beyond a manufacturer’s control, including, but not limited to the fact that the institutional review board at a given clinical site might not approve the study, might decline to renew approval which is required annually, or might suspend or terminate the study before the study has been completed. Also, the interim results of a study may not be satisfactory, leading the sponsor to terminate or suspend the study on its own initiative or the FDA may terminate or suspend the study. There is no assurance that a clinical study at any given site will progress as anticipated; there may be an insufficient number of patients who qualify for the study or who agree to participate in the study or the investigator at the site may have priorities other than the study. Also, there can be no assurance that the clinical study will provide sufficient evidence to assure the FDA that the product is safe and effective, a prerequisite for FDA approval of a PMA, or substantially equivalent in terms of safety and effectiveness to a predicate device, a prerequisite for clearance under 510(k). Even if the FDA approves or clears a device, it may limit its intended uses in such a way that manufacturing and distributing the device may not be commercially feasible. For marketing outside the U.S., we also will be subject to foreign regulatory requirements governing clinical trials and marketing approval for medical devices. The requirements governing the conduct of clinical trials, device clearance/approval, pricing, and reimbursement vary widely from country to country. In addition to the regulatory clearance and approval processes described herein, the FDA periodically issues draft guidance documents designed to provide additional detail on or reform aspects of the 510(k) and PMA clearance and approval processes. To the extent the FDA finalizes and implements these documents, the average 510(k) and PMA submission requirements and review times may change and devices that might previously have been cleared under the 510(k) process may require approval under the PMA process (and vice-versa). Additionally, since 2012, the FDA has collected user fees for the review of certain premarket submissions received on or after October 1, 2012, including 510(k) and PMA applications. These fees are intended to improve the device review process, but it is still too early to assess the actual impact on the industry.
After clearance or approval to market is given, the FDA and foreign regulatory agencies, upon the occurrence of certain events, are authorized under various circumstances to withdraw the clearance or approval or require changes to a device, its manufacturing process or its labeling or additional proof that regulatory requirements have been met.
A manufacturer of a device approved through the PMA is not permitted to make changes to the device, which affects its safety or effectiveness without first submitting a supplement application to its PMA and obtaining FDA approval for that supplement. In some instances, the FDA may require clinical trials to support a supplement application. A manufacturer of a device cleared through the 510(k) process must submit another premarket notification if it intends to make a change or modification in the device that could significantly affect the safety or effectiveness of the device, such as a significant change or modification in design, material, chemical composition, energy source or manufacturing process. Any change in the intended uses of a PMA device or a 510(k) device requires an approved PMA supplement or a cleared premarket notification. Exported devices are subject to the regulatory requirements of each country to which the device is exported, as well as certain FDA export requirements.
21
A company that intends to manufacture medical devices is required to register with the FDA before it begins to manufacture the device for commercial distribution. As a result, we and any entity that manufactures products on our behalf will be subject to periodic inspection by the FDA for compliance with the FDA’s Quality System Regulation requirements and other regulations. In the European Community, we will be required to maintain certain International Organization for Standardization (“ISO”), certifications in order to sell products and we or our manufacturers undergo periodic inspections by notified bodies to obtain and maintain these certifications. These regulations require us or our manufacturers to manufacture products and maintain documents in a prescribed manner with respect to design, manufacturing, testing and control activities. Further, we are required to comply with various FDA and other agency requirements for labeling and promotion. The Medical Device Reporting regulations require that we provide information to the FDA whenever there is evidence to reasonably suggest that a device may have caused or contributed to a death or serious injury or, if a malfunction were to occur, could cause or contribute to a death or serious injury. In addition, the FDA prohibits us from promoting a medical device for unapproved indications.
Diagnostic Products
Certain of our diagnostic products in development are subject to regulation by the FDA and similar international health authorities. For these products, we have an obligation to adhere to the FDA’s cGMP regulations. Additionally, we are subject to periodic FDA inspections, quality control procedures, and other detailed validation procedures. If the FDA finds deficiencies in the validation of our manufacturing and quality control practices, it may impose restrictions on marketing these specific products until corrected.
Regulation by governmental authorities in the U.S. and other countries may be a significant factor in how we develop, test, produce and market our diagnostic test products. Diagnostic tests like ours may not fall squarely within the regulatory approval process for pharmaceutical or device products as described above, and the regulatory pathway is not as clear. Although the FDA regulates in vitro diagnostic devices, some laboratory companies have successfully commercialized diagnostic tests for various conditions and disease states without seeking clearance or approval for such tests through a 510(k) or PMA approval process. These tests are known as laboratory developed tests (“LDTs”) and are designed, manufactured, and used within a single laboratory that is certified under the CLIA. CLIA is a federal law that regulates clinical laboratories that perform testing on specimens derived from humans for the purpose of providing information for diagnostic, preventative or treatment purpose. Such LDT testing is currently under the purview of CMS and state agencies that provide oversight of the safe and effective use of LDTs. A large number of laboratory testing in the United States consists of LDTs.
However, the FDA has consistently asserted that it has the regulatory authority to regulate LDTs despite historically exercising enforcement discretion. In furtherance of that position, the FDA issued two draft guidance documents in October 2014: (1) Framework for Regulatory Oversight of Laboratory Developed Tests (the “Framework Guidance”); and (2) FDA Notification and Medical Device Reporting for Laboratory Developed Tests (the “Notification Guidance”), but has taken no action on the draft guidance. Rather, Congress is considering various legislation that, if enacted, could formalize an FDA oversight role for LDTs, including both the Verifying Accurate Leading-edge IVCT Development (VALID) Act, and the Verified Innovative Testing in American Laboratories (VITAL) Act. The FDA has informally indicated that it is giving Congress the opportunity to develop a legislative solution.
If enacted, legislation such as the VALID Act or the VITAL Act may have a materially adverse effect on the time, cost, and risk associated with the Company’s development and commercialization of LDTs for the U.S. market, and there can be no assurance that clearances or approvals sought by the Company will be granted and maintained. However, the FDA’s authority to regulate LDTs continues to be challenged, the proposed VALID Act and VITAL Act have faced opposition, and the regulatory situation remains fluid. The FDA has indicated that it will continue dialogue with the industry, and the timeline and process for action by Congress or the FDA is unknown. We will continue to monitor changes to all domestic and international LDT regulatory policy so as to ensure compliance with the current regulatory scheme.
Impact of Regulation
The FDA in the course of enforcing the FDCA may subject a company to various sanctions for violating FDA regulations or provisions of the FDCA, including requiring recalls, issuing Warning Letters, seeking to impose civil money penalties, seizing devices that the agency believes are non-compliant, seeking to enjoin distribution of a specific drug or device seeking to revoke a clearance or approval, seeking disgorgement of profits and seeking to criminally prosecute a company and its officers and other responsible parties.
The levels of revenues and profitability of biopharmaceutical companies may be affected by the continuing efforts of government and third party payors to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing or profitability of therapeutic and other pharmaceutical products is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement
22
similar governmental control. In addition, in the U.S. and elsewhere, sales of therapeutic and other pharmaceutical products are dependent in part on the availability and adequacy of reimbursement from third party payors, such as the government or private insurance plans. Third party payors are increasingly challenging established prices, and new products that are more expensive than existing treatments may have difficulty finding ready acceptance unless there is a clear therapeutic benefit. On April 1, 2014, the Protecting Access to Medicare Act of 2014 (“PAMA”) was enacted into law. Under PAMA, Medicare payment for clinical diagnostic laboratory tests are established by calculating a weighted mean of private payor rates with new rates. Effective January 1, 2018, clinical laboratory fee schedule rates were based on weighted median private payor rates as required by PAMA. We cannot assure you that any of our products will be considered cost effective, or that reimbursement will be available or sufficient to allow us to sell them competitively and profitably.
State and Federal Security and Privacy Regulations
The privacy and security regulations under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 ( the “HITECH Act”, and collectively, “HIPAA”), establish comprehensive federal standards with respect to the uses and disclosures of protected health information, or PHI, by health plans and health care providers, in addition to setting standards to protect the confidentiality, integrity and availability of electronic PHI. The regulations establish a complex regulatory framework on a variety of subjects, including:
•the circumstances under which uses and disclosures of PHI are permitted or required without a specific authorization by the patient, including but not limited to treatment purposes, to obtain payments for services and health care operations activities;
•a patient’s rights to access, amend and receive an accounting of certain disclosures of PHI;
•the content of notices of privacy practices for PHI; and
•administrative, technical and physical safeguards required of entities that use or receive PHI electronically.
The final omnibus rule implementing the HITECH Act took effect on March 26, 2013. The rule is broad in scope, but certain provisions are particularly significant in light of our business operations. For example, the final “omnibus” rule implementing the HITECH Act:
•Makes clear that situations involving impermissible access, acquisition, use or disclosure of protected health information are now presumed to be a breach unless the covered entity or business associate is able to demonstrate that there is a low probability that the information has been compromised;
•Defines the term “business associate” to include subcontractors and agents that receive, create, maintain or transmit protected health information on behalf of the business associate;
•Establishes new parameters for covered entities and business associates on uses and disclosures of PHI for fundraising and marketing; and
•Establishes clear restrictions on the sale of PHI without patient authorization.
As a provider of clinical laboratory services and as we launch commercial diagnostic tests, we must continue to implement policies and procedures related to compliance with the HIPAA privacy and security regulations, as required by law. The privacy and security regulations provide for significant fines and other penalties for wrongful use or disclosure of PHI, including potential civil and criminal fines and penalties.
Additionally, as we operate in Europe, we may be subject to laws governing the collection, use, disclosure and transmission of personal and/or patient information. In December 2015, the European Union approved a General Data Protection Regulation (“GDPR”) to replace the current data protection directive, Directive 95/46/EC, which took effect May 25, 2018. The GDPR governs the use and transfer of personal data and imposes enhanced penalties for noncompliance. We have made, and will continue to make, certain adjustments to our operations so as to comply with the GDPR.
Anti-Kickback Laws, Physician Self-Referral Laws, False Claims Act, Civil Monetary Penalties
We are also subject to various federal, state, and international laws pertaining to health care “fraud and abuse,” including anti-kickback laws and false claims laws. The federal Anti-Kickback Statute prohibits anyone from knowingly and willfully soliciting, receiving, offering, or paying any remuneration with the intent to refer, or to arrange for the referral or order of, services or items payable under a federal health care program, including the purchase or prescription of a particular drug or the use of a service or device. Recognizing that the Anti-Kickback Statute is broad and may technically prohibit many innocuous or beneficial arrangements, Congress authorized the U.S. Department of Health and Human Services Office of Inspector General, or OIG, to issue a series of regulations, known as “safe harbors.” These safe harbors set forth requirements that, if met
23
in their entirety, will assure health care providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal, or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities, such as the OIG.
Violations of the Anti-Kickback Statute are punishable by the imposition of criminal fines, civil money penalties, treble damages, and/or exclusion from participation in federal health care programs. Many states have also enacted similar anti-kickback laws. The Anti-Kickback Statute and similar state laws and regulations are expansive. If the government were to allege against or convict us of violating these laws, there could be a material adverse effect on our business, results of operations, financial condition, and our stock price. Even an unsuccessful challenge could cause adverse publicity and be costly to respond to, which could have a materially adverse effect on our business, results of operations and financial condition. We will consult counsel concerning the potential application of these and other laws to our business and our sales, marketing and other activities and will make good faith efforts to comply with them. However, given the broad reach of federal and state anti-kickback laws and the increasing attention given by law enforcement authorities, we are unable to predict whether any of our activities will be challenged or deemed to violate these laws.
We are also subject to the physician self-referral laws, commonly referred to as the Stark law, which is a strict liability statute that generally prohibits physicians from referring Medicare patients to providers of “designated health services,” including clinical laboratories, with whom the physician or the physician’s immediate family member has an ownership interest or compensation arrangement, unless an applicable exception applies. Moreover, many states have adopted or are considering adopting similar laws, some of which extend beyond the scope of the Stark law to prohibit the payment or receipt of remuneration for the prohibited referral of patients for designated healthcare services and physician self-referrals, regardless of the source of the payment for the patient’s care. If it is determined that certain of our practices or operations violate the Stark law or similar statutes, we could become subject to civil and criminal penalties, including exclusion from the Medicare programs and loss of government reimbursement. The imposition of any such penalties could harm our business.
Another development affecting the health care industry is the increased use of the federal civil False Claims Act and, in particular, actions brought pursuant to the False Claims Act’s “whistleblower” or “qui tam” provisions. The False Claims Act, as amended by the Fraud Enforcement and Recovery Act of 2009 and the Patient Protection and Affordable Care Act of 2010 (“Affordable Care Act”), imposes liability on any person or entity who, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment by a federal health care program. We submit claims for services performed at our laboratories. The qui tam provisions of the False Claims Act allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought by private individuals has increased dramatically. In addition, various states have enacted false claim laws analogous to the False Claims Act. Many of these state laws apply where a claim is submitted to any third-party payor and not merely a federal health care program. When an entity is determined to have violated the False Claims Act, it may be required to pay up to three times the actual damages sustained by the government, plus civil penalties for each separate false claim. There are many potential bases for liability under the False Claims Act. Liability arises, primarily, when an entity knowingly submits, or causes another to submit, a false claim for reimbursement to the federal government. The False Claims Act has been used to assert liability on the basis of inadequate care, kickbacks and other improper referrals, improper use of Medicare numbers when detailing the provider of services, and allegations as to misrepresentations with respect to the services rendered. Our activities relating to the sale and marketing of our products may be subject to scrutiny under these laws. We are unable to predict whether we would be subject to actions under the False Claims Act or a similar state law, or the impact of such actions. However, the costs of defending such claims, as well as any sanctions imposed, could significantly adversely affect our financial performance.
Further, the beneficiary inducement prohibition of the federal Civil Monetary Penalty Law prohibits any entity from offering or transferring to a Medicare or Medicaid beneficiary any remuneration that the entity knows or should know is likely to influence the beneficiary’s selection of a particular provider, practitioner or supplier of Medicare or Medicaid payable items or services, including waivers of copayments and deductible amounts (or any part thereof) and transfers of items or services for free or for other than fair market value. On December 7, 2016, the OIG released amendments to the CMP. Some of the amendments may impact our business, such as allowing certain remuneration to financially needy individuals. Entities found in violation may be liable for civil monetary penalties of up to $10,000 for each wrongful act. Although we believe that our sales and marketing practices are in material compliance with all applicable federal and state laws and regulations, relevant regulatory authorities may disagree and violation of these laws, or, our exclusion from such programs as Medicaid and other governmental programs as a result of a violation of such laws, could have a material adverse effect on our business, results of operations, financial condition and cash flows.
Open Payments Program
24
With the launch of Rayaldee, part of our business is now subject to the federal Physician Payments Sunshine Act under the Affordable Care Act, and its implementing regulations, which is implemented though the physicians Open Payments Program (the “Open Payments Program”). The Open Payments Program requires manufacturers of drugs, devices, biological and medical supplies covered by Medicare, Medicaid or the Children’s Health Insurance Program, to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals. Manufacturers must also report, on an annual basis, certain ownership and investment interests held by physicians and their immediate family members and payments or other “transfers of value” made to such physician owners. A failure to report each payment, other transfer of value, or ownership/investment interest in a timely, accurate, and complete manner may result in civil monetary penalties of up to $150,000 annually. Further, the “knowing” failure to report each payment, other transfer of value, or ownership/investment interest may result in a one million dollar annual penalty. Several other states and a number of countries worldwide have adopted or are considering the adoption of similar transparency laws. Any failure by us to implement proper procedures to track and report on a timely basis transfers of value to physicians and teaching hospitals could result in substantial penalties.
Foreign Corrupt Practices Act
We are also subject to the U.S. Foreign Corrupt Practices Act (“FCPA”), which prohibits corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect their transactions and to devise and maintain an adequate system of internal accounting controls. Our international activities create the risk of unauthorized payments or offers of payments by our employees, consultants, sales agents or distributors, even though they may not always be subject to our control. We discourage these practices by our employees and agents. However, our existing safeguards and any future improvements may prove to be less than effective, and our employees, consultants, sales agents or distributors may engage in conduct for which we might be held responsible. Any failure by us to adopt appropriate compliance procedures and ensure that our employees and agents comply with the FCPA and applicable laws and regulations in foreign jurisdictions could result in substantial penalties or restrictions on our ability to conduct business in certain foreign jurisdictions.
MANUFACTURING AND QUALITY
Our current pharmaceutical manufacturing facilities are located in Waterford, Ireland, Guadalajara, Mexico, Nesher, Israel, and Banyoles, Spain. In addition to such facilities, we have entered into agreements with various third parties for the formulation and manufacture of our pharmaceutical clinical supplies. These suppliers and their manufacturing facilities must comply with FDA regulations, current good laboratory practices and current good manufacturing practices (“cGMPs”). We plan to continue to outsource the manufacturing and formulation of our clinical supplies.
The FDA and similar regulatory bodies may inspect our facilities and the facilities of those who manufacture on our behalf worldwide. If the FDA or similar regulatory bodies inspecting our facilities or the facilities of our suppliers find regulatory violations in manufacturing and quality control practices or procedures they may require us to cease partial or complete manufacturing operations until the violations are corrected. They may also impose restrictions on distribution of specific products until the violations are corrected.
Our point-of-care diagnostic system consists of a disposable test cassette and an analyzer. We prepare all necessary test reagents and assemble and package the disposable cassettes at our facility in Woburn, Massachusetts. We rely on third parties for the manufacture of the analyzer.
We are committed to providing high quality products to our customers, and we plan to meet this commitment by working diligently to continue implementing updated and improved quality systems and concepts throughout our organization.
SALES & MARKETING
Our diagnostics business includes BioReference’s almost 250-person sales and marketing team in the U.S. to drive growth and leverage new products. We have a highly specialized, field based 57-person sales and marketing team in the United States dedicated to the launch and commercialization of Rayaldee. We also have limited sales and marketing personnel in Ireland, Chile, Spain, Mexico and Israel.
HUMAN CAPITAL RESOURCES
Employees and Labor Relations
25
As of December 31, 2021, we had 5,767 full-time employees worldwide. With the exception of the employees of one of our subsidiaries, OPKO Spain, based in one of our factories in Spain, none of our employees are represented by a collective bargaining agreement. Overall, we consider our employee relations to be good.
Health and Safety
As a company in the healthcare industry, employee safety is a key focus of our leadership, communications, and training. We are required to comply with the College of American Pathologists and CLIA laboratory safety requirements in addition to OSHA regulations. With a clear leader in our EHS Manager, direction, standards of practice, training and auditing are consolidated and then disseminated to our managers, supervisors and all employees. We continually align our health and safety goals with those prescribed by applicable regulatory agencies and balance these goals with the needs of our employees. During the COVID-19 pandemic, we transitioned non-essential workers from the office to working from home, and we optimized our essential worker stations in our laboratories and other key process areas to provide for appropriate sanitation, social distancing and other appropriate measures to address the risks of the pandemic. Using guidance provided by the CDC and OSHA among other agencies, we worked to ensure proper personal protective equipment, spacing, workstation design and information support were available to our essential employees who continued working in our offices and facilities during the pandemic.
Competitive Pay and Benefits
We are committed to fair pay and we offer competitive medical benefits to all of our employees. Our U.S. health benefits package is above the competitive range for similar companies in our comparative industries and is one of the key tools we use for recruitment.
Inclusion and Diversity
We recognize the importance of and value diversity and inclusion in our workplace. As such, we have celebrated our diversity through employee and social media announcements in conjunction with company newsletters and employee events. We welcome discussions about our differences, embracing them and learning from them to move forward as a stronger, more productive organization. These differences are not limited to ethnicity or religion, but also in the way we process information and communicate with our colleagues. We are in a unique position where our workforce is already quite diverse and according to feedback from employee surveys, there is great pride and respect shared among our teams.
In addition, one of our strategic business goals is to recognize and serve diverse communities. Through BioReference, we work closely with clients in these communities by offering excellent customer service and patient care.
Talent Development
We recognize it is important that our employees are able to develop and grow their careers. We have a Head of Learning and Training whose responsibility is to enhance employee training and development as well as to ensure compliance while working in a collaborative environment. Our talent acquisition team is undergoing a transformation that is changing recruitment strategies to source from more diverse channels, which we anticipate will lead to more candidate hiring options, enhance our recruitment platform and eventually strengthen employee retention.
Code of Ethics
We have adopted a Code of Business Conduct and Ethics. We require all employees, including our principal executive officer and principal accounting officer and other senior officers and our employee directors, to read and to adhere to the Code of Business Conduct and Ethics in discharging their work-related responsibilities. Employees are required to report any conduct that they believe in good faith to be an actual or apparent violation of the Code of Business Conduct and Ethics. The Code of Business Conduct and Ethics is available on our website at http://www.OPKO.com.
Available Information
We are required to file annual, quarterly and current reports, proxy statements and other information with the SEC. Information that we file with the Securities and Exchange Commission is available at the SEC’s web-site at www.sec.gov. We also make available free of charge on or through our web site, at www.opko.com, our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after such material is electronically filed with the SEC. The information on our website is not, and shall not be deemed to be, a part hereof or incorporated into this or any of our other filings with the SEC.
ITEM 1A. RISK FACTORS.
26
You should carefully consider the risks described below, as well as other information contained in this report, including the consolidated financial statements and the notes thereto and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The occurrence of any of the events discussed below could significantly and adversely affect our business, prospects, results of operations, financial condition, and cash flows.
RISKS RELATED TO OUR BUSINESS
Our business has been, and may continue to be, affected by the coronavirus disease 2019 (COVID-19) outbreak.
The outbreak of the coronavirus disease 2019 (COVID-19) has evolved into a global pandemic, significantly affecting the U.S. and most countries around the world. The extent to which this coronavirus impacts our business and operating results will depend on future developments that are highly uncertain and cannot be accurately predicted, including new information that may emerge concerning the virus, including variants of the virus, and the actions to contain the spread of or to detect, prevent, or treat COVID-19, among others.
As a result of the demand for COVID-19 testing, the Company’s overall testing volume has increased significantly, which has positively impacted its operations. Simultaneously, however, demand for tests that comprise the Company’s core testing business has declined. Should the demand for COVID-19 PCR testing decline, whether from the introduction of new technologies, vaccines or therapies or a reduction in infection rates, our business, including our sales and operations, could be materially adversely affected. Because the demand and duration of the need for COVID-19 testing are uncertain, the Company could experience significant volatility in its results of operations if the demand for testing declines and such demand is not offset by an increase in demand for the services provided by the Company’s core testing business.
We may also experience supply chain disruptions, including shortages, delays and price increases in testing equipment and supplies as a result of global disruptions in healthcare markets, which could materially adversely impact our business. It is also possible that the Company will experience an adverse impact on cash collections as a result of the COVID-19 pandemic.
Governments have implemented travel restrictions and quarantine policies which may have a material adverse economic effect on our business. Such restrictions may present challenges in connection with our laboratory business, our ability to successfully commercialize Rayaldee, our ability to manufacture pharmaceutical products in Ireland, Mexico, Spain, Chile and Israel, and our ability to continue clinical development of our product candidates. Further, if the spread of the coronavirus pandemic continues and our operations are adversely impacted, our ability to meet performance obligations under contracts may be impacted.
COVID-19 could also disrupt our operations due to absenteeism by infected or ill members of management or other employees, or absenteeism by members of management and other employees who elect not to come to work due to the illness affecting others in our office or laboratory facilities, or due to quarantines.
The regulatory framework governing laboratories, diagnostic and pharmaceutical companies may be affected as governmental authorities divert resources to respond to the COVID-19 outbreak, which may have an unanticipated and unforeseen impact on our operations. It is possible that the timing of regulatory submissions and approvals for our products, including hGH-CTP, will be adversely impacted or delayed. With respect to our ongoing and planned clinical trials, restrictions and efforts to avoid further spread of COVID-19 may present challenges to the conduct of these trials consistent with normally applicable approaches and good clinical practice standards, and although regulators including the FDA have offered guidance applicable during the COVID-19 pandemic allowing for flexibility of standards in certain areas and alternate methods of meeting trial oversight obligations (for example, via remote monitoring), the potential impact of these challenges cannot be fully predicted at this time.
We have had a history of operating losses and may not be able to sustain profitability in the near future.
BioReference’s COVID-19 testing volume has positively impacted our profitability, but we had, prior to 2020, incurred losses since our inception. We may not continue to generate substantial revenue from COVID-19 testing as vaccine use is adopted and infection rates decline, unless such decline is offset by significant revenue generation from our other income streams. We have historically generated only limited revenue from operations and we may not generate substantial revenue from the sale of proprietary pharmaceutical products or certain of our diagnostic products for some time, if at all. Other than NGENLA (Somatrogon), which has been approved in the EU, Japan, Canada and Australia, Rayaldee is our only pharmaceutical product that has been approved for marketing in the U.S. or elsewhere. We continue to incur substantial research and development and general and administrative expenses related to our operations including our pre-clinical development activities and clinical trials. We may incur losses from our operations in the future and these losses could increase as we continue our research activities and conduct development of, and seek regulatory approvals and clearances for, our product candidates, particularly if we are unable to generate or sustain profits and cash flow from sales of Rayaldee, NGENLA,
27
or our operations at BioReference. If we are unable to generate or sustain profits and cash flow from our operations, our product candidates fail in clinical trials or do not gain regulatory approval or clearance, or if our approved products and product candidates do not achieve market acceptance, we may no longer be profitable. In particular, if we are unable to successfully commercialize Rayaldee or NGENLA, we may never generate substantial revenues from Rayaldee or NGENLA. If we are unable to obtain FDA approval for Somatrogon in the U.S., we will not be able to commercialize Somatrogon in the U.S. and will therefore not generate revenues from NGENLA in the U.S. In addition, if we are required by the FDA to perform studies in addition to those we currently anticipate, our expenses will increase beyond current expectations and the timing of any potential product approval may be delayed.
We may require additional funding, which may not be available to us on acceptable terms, or at all.
As of December 31, 2021, we had cash and cash equivalents of $134.7 million. Prior to 2020, we had not generated sustained positive cash flows sufficient to offset our operating and research and development expenses and our primary sources of cash has been from the public and private placement of stock, the issuance of convertible notes and credit facilities available to us. While we have generated significant cash from operations as a result of testing related to the COVID-19 pandemic, we are unable to predict how long the demand will continue for our COVID-19 related testing, whether pricing and reimbursement policies for testing will sustain, or whether further restrictions will be placed on elective procedures or if stay at home orders will be reinstated and accordingly, the sustainability of the cash flow is uncertain.
If we are unable to generate a sufficient amount of product and service revenue to finance our cash requirements for research, development and operations, we will need to finance future cash needs primarily through public or private equity offerings, debt financings, or strategic collaborations. Our ability to obtain additional capital may depend on prevailing economic conditions and financial, business and other factors beyond our control, as well as our ability to comply with credit facilities and other loan requirements. The amended and restated credit agreement (the “A&R Credit Agreement”) with JPMorgan Chase Bank, N.A. (“CB”) governing our revolving credit facility with CB contains, and other agreements that govern our indebtedness may contain restrictive and financial covenants that impose restrictions on us and certain of our subsidiaries, including covenants that require us to maintain specified financial ratios. We have obtained waivers and/or amended our revolving credit facility with CB from time to time in the past to avoid a default under certain covenants, and our ability to comply with these financial covenants may be adversely affected in the future. Failure to comply with specified financial covenants and other requirements could result in an event of default under our A&R Credit Agreement and/or other lenders, which, if not cured or waived, could restrict us from utilizing the facility or accelerate any repayment obligations we may have under the facility and which could have a material adverse effect on our financial condition.
Disruptions in the U.S. and global financial markets may also adversely impact the availability and cost of credit, as well as our ability to raise money in the capital markets. Economic conditions have been, and continue to be, volatile. Continued instability in these market conditions may limit our ability to replace, in a timely manner, maturing liabilities and access the capital necessary to fund and grow our business.
There can be no assurance that additional capital will be available to us on acceptable terms, or at all, which could adversely impact our business, results of operations, liquidity, capital resources and financial condition. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of, or eliminate one or more of our clinical trials or research and development programs or cease operations altogether. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional significant dilution, and debt financing, if available, may involve restrictive covenants and other onerous terms. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our products and product candidates or grant licenses on terms that may not be favorable to us.
Our research and development activities may not result in commercially viable products.
Many of our product candidates are in the early stages of development and are prone to the risks of failure inherent in drug, diagnostic, and medical device product development. These risks further include the possibility that such products would:
•be found to be ineffective, unreliable, or otherwise inadequate or otherwise fail to receive regulatory approval;
•be difficult or impossible to manufacture on a commercial scale;
•be uneconomical to market or otherwise not be effectively marketed;
•fail to be successfully commercialized if adequate reimbursement from government health administration authorities, private health insurers, and other organizations for the costs of these products is unavailable;
•be impossible to commercialize because they infringe on the proprietary rights of others or compete with products marketed by others that are superior; or
28
•fail to be commercialized prior to the successful marketing of similar products by competitors.
The results of pre-clinical trials and previous clinical trials for our products may not be predictive of future results, and our current and planned clinical trials may not satisfy the requirements of the FDA or other non-U.S. regulatory authorities.
Positive results from pre-clinical studies and early clinical trial experience should not be relied upon as evidence that later-stage or large-scale clinical trials will succeed. Likewise, there can be no assurance that the results of studies conducted by collaborators or other third parties will be viewed favorably or are indicative of our own future study results. We may be required to demonstrate with substantial evidence through well-controlled clinical trials that our product candidates are either (i) with respect to drugs or Class III devices, safe and effective for use in a diverse population for their intended uses or (ii) with respect to Class I or Class II devices, are substantially equivalent in terms of safety and effectiveness to devices that are already marketed under section 510(k) of the Food, Drug and Cosmetic Act. Success in early clinical trials does not mean that future clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and other non-U.S. regulatory authorities despite having progressed through initial clinical trials.
Further, our drug candidates may not be approved or cleared even if they achieve their primary endpoints in phase 3 clinical trials or registration trials. In addition, our diagnostic test candidates may not be approved or cleared, as the case may be, even though clinical or other data are, in our view, adequate to support an approval or clearance. The FDA or other non-regulatory authorities may disagree with our trial design and our interpretation of data from pre-clinical studies and clinical trials. In addition, any of these regulatory authorities may change requirements for the approval or clearance of a product candidate even after reviewing and providing comment on a protocol for a pivotal clinical trial that has the potential to result in FDA and other non-U.S. regulatory authorities’ approval. Any of these regulatory authorities may also approve or clear a product candidate for fewer or more limited indications or uses than we request or may grant approval or clearance contingent on the performance of costly post-marketing clinical trials. The FDA or other non-U.S. regulatory authorities may not approve the labeling claims necessary or desirable for the successful commercialization of our product candidates.
The results of our clinical trials may show that our product candidates may cause undesirable side effects, which could interrupt, delay or halt clinical trials, resulting in the denial of regulatory approval by the FDA and other non-U.S. regulatory authorities.
We rely on licensing agreements with Vifor, Nicoya, and international partners for the international development and marketing of Rayaldee. Failure to maintain these license agreements could prevent us from successfully developing and commercializing Rayaldee worldwide.
In May 2016, EirGen, our wholly-owned subsidiary, partnered with VFMCRP through a Development and License Agreement for the development and marketing of Rayaldee in Europe, Canada, Mexico, Australia, South Korea and certain other international markets. The license to VFMCRP potentially covers all therapeutic and prophylactic uses of the product in human patients, provided that initially the license is for the use of the product for the treatment or prevention of secondary hyperparathyroidism related to patients with stage 3 or 4 chronic kidney disease and vitamin D insufficiency/deficiency. Effective May 5, 2020, we entered into the VFMCRP Amendment, pursuant to which the parties agreed to exclude Mexico, South Korea, the Middle East and all of the countries of Africa from the VFMCRP Territory. In May 2021, we further amended the VFMCRP Agreement for VFMCRP to assume all the rights to Rayaldee in Japan that had been previously granted to JT. In addition, the parties agreed to certain amendments to the milestone structure and to reduce minimum royalties payable. As revised, the Company is eligible to receive up to $17 million in regulatory milestones and $210 million in milestone payments tied to launch, pricing and sales of Rayaldee, and tiered, double-digit royalties. The success of the Development and License Agreement with VFMCRP is dependent in part on, among other things, the skills, experience and efforts of VFMCRP’s employees responsible for the project, VFMCRP’s commitment to the arrangement, and the financial condition of VFMCRP, all of which are beyond our control. In the event that VFMCRP, for any reason, including but not limited to early termination of the agreement, fails to devote sufficient resources to successfully develop and market Rayaldee internationally, our ability to earn milestone payments or receive royalty payments would be adversely affected, which would have a material adverse effect on our financial condition and prospects.
In October 2017, we entered into a Development and License Agreement (the “JT Agreement”) with JT under which JT was granted the exclusive rights for the development and commercialization of Rayaldee in Japan. The JT Agreement was terminated in May 2021.
On June 18, 2021, EirGen and Nicoya entered into the Nicoya Agreement granting Nicoya the exclusive rights for the development and commercialization of (the Nicoya Product in the Nicoya Territory. The license grant to Nicoya covers the therapeutic and preventative use of the Nicoya Product for SHPT in non-dialysis and hemodialysis chronic kidney disease patients. EirGen received an initial upfront payment of $5 million and is eligible to receive an additional $5 million upon the
29
first to occur of (A) a predetermined milestone and (B) the first anniversary of the effective date. EirGen is also eligible to receive up to an additional aggregate amount of $115 million upon the achievement of certain development, regulatory and sales-based milestones by Nicoya for the Nicoya Product in the Nicoya Territory. EirGen will also receive tiered, double digit royalty payments at rates in the low double digits on net product sales within the Nicoya Territory and in the Nicoya Field. Nicoya will, at its sole cost and expense, be responsible for performing all development activities necessary to obtain all regulatory approvals for the Nicoya Product in the Nicoya Territory and for all commercial activities pertaining to the Nicoya Product in the Nicoya Territory. The success of the Nicoya Agreement is dependent in part on, Nicoya’s commitment to the product and our collaboration, as well as the experience of its employees, all of which are beyond our control.
Our exclusive worldwide agreement with Pfizer is important to our business. If we do not successfully develop Somatrogon (hGH- CTP)and/or Pfizer does not successfully commercialize Somatrogon (hGH-CTP ), our business could be adversely affected.
In December 2014, we entered into a development and commercialization agreement with Pfizer relating to our long-acting hGH- CTP for the treatment of GHD in adults and children (the “Pfizer Agreement”). Under the Pfizer Agreement, we are eligible to receive up to $275 million upon the achievement of certain regulatory milestones. Upon the launch of Somatrogon (hGH-CTP) for pediatric GHD, we are eligible to receive a regional, tiered gross profit share based upon sales of both Somatrogon and Pfizer’s Genotropin® (somatropin). We are responsible for the development program and are obligated to pay for the development up to an agreed cap, which has been exceeded. In May 2020, we entered into an Amended and Restated Development and Commercialization License Agreement (the “Restated Pfizer Agreement”) with Pfizer, effective January 1, 2020, pursuant to which the parties agreed, among other things, to share all costs for Manufacturing Activities, as defined in the Restated Pfizer Agreement, for developing a licensed product for the three indications included in the Agreement. While hGH-CTP has been approved in the EU, Japan, Canada and Australia under the name NGENLA, Pfizer received a Complete Response Letter from the FDA in January 2022 in response to the BLA we and Pfizer submitted in 2020. We and Pfizer are evaluating the FDA’s comments and will work with the agency to determine an appropriate path forward. In the event that the parties are able to obtain regulatory approvals to market a product covered by the Pfizer Agreement, we will be substantially dependent on Pfizer for the successful commercialization of such product. The success of the collaboration arrangement with Pfizer is dependent in part on, among other things, the skills, experience and efforts of Pfizer’s employees responsible for the project and Pfizer’s commitment to the arrangement. The Pfizer Agreement is terminable for any reason by Pfizer upon ninety days written notice to OPKO. In the event that Pfizer terminates the Agreement or fails to devote sufficient resources to successfully develop and commercialize any product resulting from the collaboration arrangement, our ability to earn milestone payments or receive royalty or profit sharing payments would be adversely affected, which would have a material adverse effect on our financial condition and prospects and the trading prices of our securities.
Our business is substantially dependent on our ability to achieve regulatory approval for the marketing of Somatrogon (hGH-CTP) in pediatric and adult patients and the commercial success of this product.
On October 21, 2019, we and Pfizer announced that the global phase 3 trial evaluating hGH-CTP (Somatrogon) dosed once-weekly in pre-pubertal children with GHD met its primary endpoint of non-inferiority to daily Genotropin® (somatropin) for injection, as measured by annual height velocity at 12 months. In addition, change in height standard deviation scores at six and 12 months, key secondary endpoints, were higher in the hGH-CTP dosed once-weekly cohort in comparison to the Genotropin® (somatropin) dosed once-daily cohort. hGH-CTP was generally well tolerated in this study and comparable to Genotropin® (somatropin) dosed once-daily with respect to the types, numbers and severity of the adverse events observed between the treatment arms. Although the primary endpoint and key secondary endpoints were met and the safety profile for hGH-CTP was consistent with that observed with those treated with Genotropin® (somatropin), further testing and analysis, other clinical trials or patient use may undermine those determinations or unexpected side effects may arise. We previously announced topline data from an earlier phase 3, double blind, placebo controlled study of hGH-CTP in adults with GHD. Although there was no statistically significant difference between hGH-CTP and placebo on the primary endpoint of change in trunk fat mass from baseline to 26 weeks, after unblinding the study, we identified an exceptional value of trunk fat mass reduction in the placebo group that may have affected the primary outcome. We completed post-hoc sensitivity analyses for the adult study to evaluate the influence of outliers on the primary endpoint results using multiple statistical approaches. Analyses that excluded outliers showed a statistically significant difference between hGH-CTP and placebo on the change in trunk fat mass. Additional analyses that did not exclude outliers showed mixed results. There can be no assurance that the FDA or regulatory agencies in other countries will consider the sensitivity analysis or consider the product for approval for adults with GHD.
In January 2021, we and Pfizer announced that the FDA had accepted for filing the BLA submission for the pediatric indication which was submitted in October 2020. In January 2022, Pfizer received a Complete Response Letter with respect to
30
the pediatric indication. Pfizer and the Company are evaluating the best path forward for hGH-CTP in the U.S. but there can be no assurances that we will receive an approval for hGH-CTP for the treatment of pediatric GHD by the FDA.
There can be no assurance that a BLA will be submitted for the adult indication or that we will obtain marketing approval for either the pediatric or adult indication. Before they can be marketed, our products in development must be approved by the FDA or similar foreign governmental agencies. The process for obtaining FDA marketing approval is both time-consuming and costly, with no certainty of a successful outcome. If we are unable to achieve regulatory approval for hGH-CTP to treat pediatric patients or adults with GHD, our business will be significantly adversely impacted, which could have a materially adverse effect on our business, financial condition and results of operations.
Japan’s Ministry of Health, Labour and Welfare approved NGENLA (Somatrogon) for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. In October 2021, Health Canada approved NGENLA for the long-term treatment of pediatric patients who have GHD, and Australia’s Therapeutic Goods Administration (TGA) approved NGENLA for the long-term treatment of pediatric patients with growth disturbance. NGENLA may fail to be successfully commercialized in these territories which would adversely impact our anticipated milestone payments under the Restated Pfizer Agreement and negatively affect our business, financial condition and results of operations.
Protein therapeutics have the potential to cause an immune or antibody response in patients.
Antibodies may be transient or persistent and can have no effect or can neutralize the therapeutic effect of the protein. Antibodies that neutralize the activity of a therapeutic protein are known as neutralizing antibodies. As previously reported, low titers of anti-hGH-CTP non-neutralizing antibodies were noted over a four year period in 17 subjects, or approximately 35% of the subjects, in our phase 2 open label extension study in children with GHD. The low titer non-neutralizing antibodies did not affect growth parameters or IGF-1 levels in the patients. Immunogenicity testing and analysis for our phase 3 study is ongoing, and we expect that the full results of the study will be submitted for presentation at a future scientific meeting. The FDA reviews information on immune responses observed during clinical studies and the implications on safety and efficacy and could request additional studies or analyses of hGH-CTP or could decline to approve hGH-CTP for the indications we seek. Any of these occurrences could have a material adverse impact on our business, results of operation and financial condition.
Consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals, patients treated with NGENLA may develop antibodies to somatrogon. Antibodies may be transient or persistant and can have no effect or can neutralize the therapeutic effect of the protein. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Somatrogon in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Our business is dependent on our ability to develop, launch and generate revenue from our diagnostic products.
Our business is dependent on our ability to successfully commercialize our diagnostic products. We are committing significant resources to the development and commercialization of these products, and there is no guarantee that we will be able to successfully commercialize these tests. We have limited experience in developing, manufacturing, selling, marketing and distributing innovative diagnostic tests. If we are not able to successfully develop, market or sell diagnostic tests we develop for any reason, including the failure to obtain any required regulatory approvals, or obtain favorable reimbursement, we will not generate any meaningful revenue from the sale of such tests.
Our business is substantially dependent on our ability to generate profits and cash flow from our laboratory operations.
We have made a significant investment in our laboratory operations through the acquisition of BioReference. We compete in the clinical laboratory market primarily on the basis of the quality of testing, reporting and information systems, reputation in the medical community, the pricing of services and ability to employ qualified personnel. Our failure to successfully compete on any of these factors could result in the loss of clients and a reduction in our revenues and profits. To offset efforts by payors to reduce the cost and utilization of clinical laboratory services, we will need to obtain and retain new clients and business partners and grow the laboratory operations. In response to the global pandemic, BioReference has been conducting a substantial amount of COVID-19 testing that has positively impacted our revenues. Simultaneously, however, the volume of its core testing business has decreased as a result of COVID-19. A significant reduction in COVID-19 tests ordered, specimens submitted by existing clients, or payment rates, without offsetting growth in our core business testing or client base,
31
would impact our ability to successfully maintain the growth our business has experienced and could have a material adverse impact on our ability to generate profits and cash flow from the laboratory operations in the future.
Discontinuation or recalls of existing testing products, failure to develop, or acquire, licenses for new or improved testing technologies or our clients using new technologies to perform their own tests could adversely affect our business.
From time to time, manufacturers discontinue or recall reagents, test kits or instruments used by us to perform laboratory testing. Such discontinuations or recalls could adversely affect our costs, testing volume and revenue.
The clinical laboratory industry is subject to changing technology and new product introductions. Our success in maintaining a leadership position in genomic and other advanced testing technologies will depend, in part, on our ability to develop, acquire or license new and improved technologies on favorable terms and to obtain appropriate coverage and reimbursement for these technologies. We may not be able to negotiate acceptable licensing arrangements and it cannot be certain that such arrangements will yield commercially successful diagnostic tests. If we are unable to license these testing methods at competitive rates, our research and development costs may increase as a result. In addition, if we are unable to license or develop new or improved technologies to expand our esoteric testing operations, our testing methods may become outdated when compared with our competition and testing volume and revenue may be materially and adversely affected.
Currently, most clinical laboratory testing is categorized as “high” or “moderate” complexity, and thereby is subject to extensive and costly regulation under CLIA. The cost of compliance with CLIA makes it impractical for most physicians to operate clinical laboratories in their offices, and other laws limit the ability of physicians to have ownership in a laboratory and to refer tests to such a laboratory. Manufacturers of laboratory equipment and test kits could seek to increase their sales by marketing point-of-care laboratory equipment to physicians and by selling test kits approved for home or physician office use to both physicians and patients. Diagnostic tests approved for home use are automatically deemed to be “waived” tests under CLIA and may be performed in physician office laboratories as well as by patients in their homes with minimal regulatory oversight. Other tests meeting certain FDA criteria also may be classified as “waived” for CLIA purposes. The FDA has regulatory responsibility over instruments, test kits, reagents and other devices used by clinical laboratories and has taken responsibility from the Centers for Disease Control for classifying the complexity of tests for CLIA purposes. Increased approval of “waived” test kits could lead to increased testing by physicians in their offices or by patients at home, which could affect our market for laboratory testing services and negatively impact our revenues. If our competitors develop and market products that are more effective, safer or less expensive than our products and product candidates, our net revenues, profitability and commercial opportunities will be negatively impacted.
If our competitors develop and market products or services that are more effective, safer or less expensive than our current and future products or services, our revenues, profitability and commercial opportunities will be negatively impacted.
Numerous companies, including major pharmaceutical companies, specialty pharmaceutical companies and specialized biotechnology companies, are engaged in the development, manufacture and marketing of pharmaceutical products competitive with those that we intend to commercialize ourselves and through our partners. Competitors to our diagnostics business include major diagnostic companies, reference laboratories, molecular diagnostic firms, universities and research institutions. Most of these companies have substantially greater financial and other resources, larger research and development staffs and more extensive marketing and manufacturing organizations than ours. This enables them, among other things, to make greater research and development investments and efficiently utilize their research and development costs, as well as their marketing and promotion costs, over a broader revenue base. This also provides our competitors with a competitive advantage in connection with the highly competitive product acquisition and product in-licensing process. Our competitors may also have more experience and expertise in obtaining marketing approvals from the FDA and other regulatory authorities. We cannot predict with accuracy the timing or impact of the introduction of potentially competitive products or their possible effect on our sales. In addition to product development, testing, approval, and promotion, other competitive factors in the pharmaceutical and diagnostics industry include industry consolidation, product quality and price, product technology, reputation, customer service, and access to technical information.
The clinical laboratory business is intensely competitive both in terms of price and service. Pricing of laboratory testing services is often one of the most significant factors used by health care providers and third-party payors in selecting a laboratory. As a result of the clinical laboratory industry undergoing significant consolidation, larger clinical laboratory providers are able to increase cost efficiencies afforded by large-scale automated testing. This consolidation results in greater price competition. We may be unable to increase cost efficiencies sufficiently, if at all, and as a result, our net earnings and cash flows could be negatively impacted by such price competition. Additionally, we may also face changes in contracting with third party payors, fee schedules, competitive bidding for laboratory services or other actions or pressures reducing payment schedules as a result of increased or additional competition.
32
If our competitors market products that are more effective, safer, easier to use or less expensive than our current products and product candidates, or that reach the market sooner than our products and product candidates, we may not achieve commercial success. In addition, the biopharmaceutical, diagnostic, medical device, and laboratory industries are characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies, products or product candidates obsolete or less competitive.
Our product development activities could be delayed or stopped.
We do not know whether our current or planned pre-clinical and clinical studies will be completed on schedule, or at all. Furthermore, we cannot guarantee that our planned pre-clinical and clinical studies will begin on time or at all. The commencement of our planned clinical trials could be substantially delayed or prevented by several factors, including:
•a limited number of, and competition for, suitable patients with the particular types of disease required for enrollment in our clinical trials or that otherwise meet the protocol’s inclusion criteria and do not meet any of the exclusion criteria;
•a limited number of, and competition for, suitable serum or other samples from patients with particular types of disease required for our validation studies;
•a limited number of, and competition for, suitable sites to conduct our clinical trials;
•delay or failure to obtain FDA or other non-U.S. regulatory authorities’ approval or agreement to commence a clinical trial;
•delay or failure to obtain sufficient supplies of the product candidate for our clinical trials;
•requirements to provide the drugs, diagnostic tests, or medical devices required in our clinical trial protocols or clinical trials at no cost or cost, which may require significant expenditures that we are unable or unwilling to make;
•delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or investigators;
•delay or failure to obtain institutional review board (“IRB”) approval to conduct or renew a clinical trial at a prospective site; and
•insufficient liquidity to fund our preclinical and clinical studies.
The completion of our clinical trials could also be substantially delayed or prevented by several factors, including:
•slower than expected rates of patient recruitment and enrollment;
•failure of patients to complete the clinical trial;
•unforeseen safety issues;
•lack of efficacy evidenced during clinical trials;
•termination of our clinical trials by one or more clinical trial sites;
•inability or unwillingness of patients or medical investigators to follow our clinical trial protocols;
•inability to monitor patients adequately during or after treatment; and
•insufficient liquidity to fund ongoing studies.
Our clinical trials may be suspended or terminated at any time by the FDA, other regulatory authorities, the IRB for any given site, or us. Additionally, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes with appropriate regulatory authorities. Amendments may require us to resubmit our clinical trial protocols to IRBs for re-examination, which may impact the costs, timing, or successful completion of a clinical trial. Any failure or significant delay in commencing or completing clinical trials for our product candidates could materially harm our results of operations and financial condition, as well as the commercial prospects for our product candidates.
Our inability to meet regulatory quality standards applicable to our manufacturing and quality processes and to address quality control issues in a timely manner could delay the production and sale of our products or result in recalls of products.
33
Manufacturing or design defects, unanticipated use of our products, or inadequate disclosure of risks relating to the use of our products could lead to injury or other adverse events. These events could lead to recalls or safety alerts relating to our products (either voluntary or required by governmental authorities) and could result, in certain cases, in the removal of a product from the market. Any recall could result in significant costs as well as negative publicity that could reduce demand for our products. Personal injuries relating to the use of our products can also result in product liability claims being brought against us. In some circumstances, such adverse events could also cause delays in new product approvals.
We are committed to providing high quality products to our customers, and we plan to meet this commitment by working diligently to continue implementing updated and improved quality systems and concepts throughout our organization. We cannot assure you that we will not have quality control issues in the future, which may result in warning letters and citations from the FDA. If we receive any warning letters from the FDA in the future, there can be no assurances regarding the length of time or cost it will take us to resolve such quality issues to our satisfaction and to the satisfaction of the FDA. If our remedial actions are not satisfactory to the FDA, we may have to devote additional financial and human resources to our efforts, and the FDA may take further regulatory actions against us including, but not limited to, assessing civil monetary penalties or imposing a consent decree on us, which could result in further regulatory constraints, including the governance of our quality system by a third party. Our inability to resolve these issues or the taking of further regulatory action by the FDA may weaken our competitive position and have a material adverse effect on our business, results of operations and financial condition.
We manufacture pharmaceutical products in Ireland, Mexico, Spain, and Israel. We also prepare necessary test reagents and assemble and package the cassettes for our point-of-care diagnostic system at our facility in Woburn, Massachusetts. Any quality control issues at our facilities may weaken our competitive position and have a material adverse effect on our business results of operations and financial condition.
As a medical device manufacturer, we are required to register with the FDA and are subject to periodic inspection by the FDA for compliance with its Quality System Regulation (“QSR”) requirements, which require manufacturers of medical devices to adhere to certain regulations, including testing, quality control and documentation procedures. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. In addition, most international jurisdictions have adopted regulatory approval and periodic renewal requirements for medical devices, and we must comply with these requirements in order to market our products in these jurisdictions. In the European Community, we are required to maintain certain ISO certifications in order to sell our products and must undergo periodic inspections by notified bodies to obtain and maintain these certifications. Further, some emerging markets rely on the FDA’s Certificate for Foreign Government (“CFG”) in lieu of their own regulatory approval requirements. Our failure, or our manufacturers’ failure to meet QSR, ISO, or any other regulatory requirements or industry standards could delay production of our products and lead to fines, difficulties in obtaining regulatory clearances, recalls or other consequences, which could, in turn, have a material adverse effect on our business, results of operations, and our financial condition.
Failure to establish, and perform to, appropriate quality standards to assure that the highest level of quality is observed in the performance of our testing services could adversely affect the results of our operations and adversely impact our reputation.
The provision of clinical testing services, including anatomic pathology services, and related services, and the design, manufacture and marketing of diagnostic products involve certain inherent risks. The services that we provide and the products that we design, manufacture and market are intended to provide information for healthcare providers in providing patient care. Therefore, users of our services and products may have a greater sensitivity to errors than the users of services or products that are intended for other purposes.
Similarly, negligence in performing our services can lead to injury or other adverse events. We may be sued under physician liability or other liability law for acts or omissions by our pathologists, laboratory personnel and other employees. We are subject to the attendant risk of substantial damages awards and risk to our reputation.
Even after we receive regulatory approval or clearance to market our product candidates, the market may not be receptive to our products.
Our products may not gain market acceptance among physicians, patients, health care payors and/or the medical community. We believe that the degree of market acceptance will depend on a number of factors, including:
•timing of market introduction of competitive products;
•safety and efficacy of our product compared to other products;
•prevalence and severity of any side effects;
34
•potential advantages or disadvantages over alternative treatments;
•strength of marketing and distribution support;
•price of our products, both in absolute terms and relative to alternative treatments;
•availability of coverage and reimbursement from government and other third-party payors;
•potential product liability claims;
•limitations or warnings contained in a product’s regulatory authority-approved labeling; and
•changes in the standard of care for the targeted indications for any of our products or product candidates, which could reduce the marketing impact of any claims that we could make following applicable regulatory authority approval.
In addition, our efforts to educate the medical community and health care payors on the benefits of our products and product candidates may require significant resources and may never be successful. If our products do not gain market acceptance, it would have a material adverse effect on our business, results of operations, and financial condition.
If our products are not covered and eligible for reimbursement from government and third party payors, we may not be able to generate significant revenue or achieve or sustain profitability.
The coverage and reimbursement status of newly approved or cleared drugs, diagnostic and laboratory tests is uncertain, and failure of our pharmaceutical products, diagnostic tests or laboratory tests to be adequately covered by insurance and eligible for adequate reimbursement could limit our ability to market any future product candidates we may develop and decrease our ability to generate revenue from any of our existing and future product candidates that may be approved or cleared. The commercial success of our existing and future products in both domestic and international markets will depend in part on the availability of coverage and adequate reimbursement from third-party payors, including government payors, such as the Medicare and Medicaid programs, managed care organizations, and other third-party payors, as well as our ability to obtain in network status with such payors. The government and other third-party payors are increasingly attempting to contain health care costs by limiting both insurance coverage and the level of reimbursement for new drugs and diagnostic tests and restricting in network status of laboratory providers. As a result, they may not cover or provide adequate payment for our product candidates. These payors may conclude that our products are less safe, less effective, or less cost-effective than existing or later-introduced products. These payors may also conclude that the overall cost of the procedure using one of our devices exceeds the overall cost of the competing procedure using another type of device, and third-party payors may not approve our products for insurance coverage and adequate reimbursement or approve our laboratory for in network status.
The failure to obtain adequate coverage or any reimbursement for our products, or health care cost containment initiatives that limit or restrict reimbursement for our products, may reduce any future product revenue. Even though a drug (not administered by a physician) may be approved by the FDA, this does not mean that a Prescription Drug Plan (“PDP”), a private insurer operating under Medicare Part D, will list that drug on its formulary or will set a reimbursement level. PDPs are not required to make every FDA-approved drug available on their formularies. If our drug products are not listed on sufficient number of PDP formularies or if the PDPs’ levels of reimbursement are inadequate, our business, results of operations and financial condition could be materially adversely affected. Private health plans, such as managed care plans and pharmacy benefit management (“PBM”) programs may also not include our products on formularies, and may use other techniques that restrict access to our products or set a lower reimbursement rate than anticipated.
A significant portion of our revenues come from government subsidized healthcare programs such as Medicaid and Medicare. Our failure to comply with applicable Medicare, Medicaid and other governmental payor rules could result in our inability to participate in a governmental payor program, our returning funds already paid to us, civil monetary penalties, criminal penalties and/or limitations on the operational function of our laboratory.
If we were unable to receive reimbursement under a governmental payor program, a substantial portion of our consolidated revenues would be lost, which would adversely affect our results of operations and financial condition. In addition, if a federal government shutdown were to occur for a prolonged period of time, federal government payment obligations, including its obligations under Medicaid and Medicare, may be delayed. Similarly, if state government shutdowns were to occur, state payment obligations may be delayed. If the federal or state governments fail to make payments under these programs on a timely basis, our business could suffer, and our financial position, results of operations or cash flows may be materially affected.
35
As we evolve from a company primarily involved in development to a company also involved in commercialization of our pharmaceutical and diagnostic products, as well as our laboratory testing services, we may encounter difficulties in managing our growth and expanding our operations successfully.
As we advance our product candidates and expand our business, we will need to expand our development, regulatory and commercial infrastructure. As our operations expand, we expect that we will need to manage additional relationships with various third parties, collaborators and suppliers. Maintaining these relationships and managing our future growth will impose significant added responsibilities on members of our management. We must be able to: manage our development efforts and operations effectively; manage our clinical trials effectively; hire, train and integrate additional management, administrative and sales and marketing personnel; improve our managerial, development, operational and finance systems; implement and manage an effective marketing strategy; and expand our facilities, all of which may impose a strain on our administrative and operational infrastructure.
Our success is dependent to a significant degree upon the involvement, efforts and reputation of our Chairman and Chief Executive Officer, Phillip Frost, M.D.
Our success is dependent to a significant degree upon the efforts of our Chairman and CEO, Phillip Frost, M.D., who is essential to our business. The departure of our CEO for whatever reason or the inability of our CEO to continue to serve in his present capacity could have a material adverse effect upon our business, financial condition and results of operations. Our CEO has a highly regarded reputation in the pharmaceutical and medical industry and attracts business opportunities and assists both in negotiations with acquisition targets, investment targets and potential joint venture partners. Our CEO has also provided financing to us, both in terms of a credit agreement and equity investments. If we lost his services or if his reputation was damaged for whatever reason, including, but not limited to, as a result of the allegations underlying various past SEC and shareholder lawsuits against us and Dr. Frost, our relationships with acquisition and investment targets, joint ventures, customers and investors, as well as our ability to obtain additional funding on acceptable terms, or at all, may suffer and could cause a material adverse impact on our operations, financial condition and the value of our Common Stock.
If we fail to attract and retain key management and scientific personnel, we may be unable to successfully operate our business and develop or commercialize our products and product candidates.
We will need to expand and effectively manage our managerial, operational, sales, financial, development, and other resources in order to successfully operate our business and pursue our research, development, and commercialization efforts for our products and product candidates. Our success depends on our continued ability to attract, retain, and motivate highly qualified management and pre-clinical and clinical personnel. The loss of the services or support of any of our senior management could delay or prevent the development and commercialization of our products and product candidates.
Business combinations may disrupt our business, distract our management, may not proceed as planned, and may also increase the risk of potential third party claims and litigation.
One aspect of our business strategy calls for acquisitions of businesses and assets that complement or expand our current business and potential disposition of assets and businesses that may no longer help us meet our objectives, which may present greater risks for us than those faced by peer companies that do not consider acquisitions or dispositions as a part of their business strategy. We may not be able to identify attractive acquisition opportunities or, when we decide to sell assets or a business, we may encounter difficulty in finding buyers or alternative exit strategies on acceptable terms in a timely manner, or at all. Even if we do identify attractive opportunities, we or the buyer may not be able to complete the acquisition due to financing or other market constraints. If we acquire an additional business, we could have difficulty integrating its operations, systems, management and other personnel and technology with our own. There may also be unasserted claims or assessments that we failed or were unable to discover or identify in the course of performing due diligence investigations of target businesses, resulting in a loss of value. Dispositions may increase our exposure to third parties claims or litigation that may require expenditure of additional resources or negatively affect the successful outcome of the disposition. Dispositions may also involve continued financial involvement in the divested business, such as through guarantees, indemnities or other financial obligations. Under these arrangements, performance by the divested businesses or other conditions outside of our control could affect our future financial results. Moreover, seeking acquisition and divestiture opportunities and evaluating and completing them require significant investment of time and resources, may disrupt the Company’s business and distract management’s attention from day-to-day business operations.
We may fail to realize the anticipated benefits of the sale of GeneDx and we could recognize an impairment charge in the future, depending on Sema4’s stock price when the transaction closes.
36
In January 2022, Sema4 and OPKO announced they have signed the GeneDx Merger Agreement, pursuant to which Sema4 will acquire GeneDx for an upfront payment of $150 million in cash, together with 80.0 million shares of Sema4 Common Stock, subject to a customary purchase price adjustment mechanism. Additionally, Sema4 agreed to pay OPKO up to an additional $150.0 million, which may be paid in Sema4 Common Stock, cash or a combination thereof in Sema4’s discretion, subject to GeneDx achieving certain revenue targets for the fiscal years ending December 31, 2022 and 2023. Based on the closing stock price of Sema4 Common Stock as of January 14, 2022, the total upfront consideration is approximately $473 million, and the total aggregate consideration including potential milestones is approximately $623 million. If GeneDx is not able to successfully achieve its growth objectives, some of the anticipated benefits of the sale may not be realized fully.
As of December 31, 2021, GeneDx met the held-for-sale accounting criteria and the related assets and liabilities are
classified as held for sale in the consolidated balance sheet. Depending upon the value Sema4 shares upon closing of the transaction, an impairment charge may be incurred.
If the FDA or other applicable regulatory authorities approve generic products that compete with any of our products or product candidates, the sale of our products or product candidates may be adversely affected.
Once an NDA is approved, the product covered thereby becomes a “listed drug” which, in turn can be relied upon by potential competitors in support of an approval of an abbreviated new drug application, or ANDA, or 505(b)(2) application. U.S. laws and other applicable policies provide incentives to manufacturers to create modified, non-infringing versions of a drug to facilitate the approval of an ANDA or other application for a generic substitute. These manufacturers might only be required to conduct a relatively inexpensive study to show that their product has the same active ingredient(s), dosage form, strength, route of administration, and conditions of use, or labeling, as our product or product candidate and that the generic product is bioequivalent to ours, meaning it is absorbed in the body at the same rate and to the same extent as our product or product candidate. These generic equivalents, which must meet the same quality standards as branded pharmaceuticals, would be significantly less costly than ours to bring to market and companies that produce generic equivalents are generally able to offer their products at lower prices. Thus, after the introduction of a generic competitor, a significant percentage of sales of any branded product is typically lost to the generic product. Accordingly, competition from generic equivalents to our products or product candidates would materially adversely impact our revenues, profitability and cash flows and substantially limit our ability to obtain a return on the investments that we have made in our products and product candidates.
We rely on third parties to manufacture and supply our pharmaceutical and diagnostic products and product candidates.
If our manufacturing partners are unable to produce our products in the amounts that we require, we may not be able to establish a contract and obtain a sufficient alternative supply from another supplier on a timely basis and in the quantities we require. We expect to continue to depend on third-party contract manufacturers for the foreseeable future.
Our products and product candidates require precise, high quality manufacturing. Any of our contract manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and other non-U.S. regulatory authorities to ensure strict compliance with QSR regulations for devices or cGMPs for drugs, and other applicable government regulations and corresponding standards relating to matters such as testing, quality control, and documentation procedures. If our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with QSR or cGMPs, we may experience manufacturing errors resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our products, cost overruns, or other problems that could seriously harm our business.
Any performance failure on the part of our contract manufacturers could delay clinical development or regulatory approval or clearance of our product candidates or commercialization of our products and product candidates, depriving us of potential product revenue and resulting in additional losses. In addition, our dependence on a third party for manufacturing may adversely affect our future profit margins. Our ability to replace an existing manufacturer may be difficult because the number of potential manufacturers is limited and the FDA must approve any replacement manufacturer before it can begin manufacturing our products or product candidates. Such approval would result in additional non-clinical testing and compliance inspections. It may be difficult or impossible for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or at all.
Independent clinical investigators and contract research organizations that we engage to conduct our clinical trials may not be diligent, careful or timely.
We depend on independent clinical investigators to conduct our clinical trials. Contract research organizations may also assist us in the collection and analysis of data. These investigators and contract research organizations are independent
37
contractors and we will not be able to control, other than by contract, the amount of resources, including time, that they devote to products that we develop. If independent investigators fail to devote sufficient resources to the development of product candidates or clinical trials, or if their performance is substandard, it will delay the marketing approval or clearance and commercialization of any products that we develop. Further, the FDA requires that we comply with standards, commonly referred to as good clinical practice, for conducting, recording and reporting clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity, and confidentiality of trial subjects are protected. If our independent clinical investigators and contract research organizations fail to comply with good clinical practice, the results of our clinical trials could be called into question and the clinical development of our product candidates could be delayed.
Failure of clinical investigators or contract research organizations to meet their obligations to us or comply with federal regulations and good clinical practice procedures could adversely affect the clinical development of our product candidates and harm our business, results of operations, and financial condition.
If the validity of an informed consent from a subject was to be challenged, it may negatively impact our product development efforts.
We take steps to ensure that all clinical data and genetic and other biological samples are collected from subjects who provide informed consent for the data and samples as required by applicable laws and we work to ensure that the subjects from whom our data and samples are collected do not retain any proprietary or commercial rights to the data or samples or any discoveries derived from them. However, because we may collect data and samples from countries that are governed by a number of different regulatory regimes, there are many complex legal questions relating to the adequacy of informed consent that we must continually address. The adequacy of any given subject’s informed consent may be challenged in the future, and any given informed consent may prove unlawful or otherwise inadequate for our purposes. Any findings against us, or our clinical collaborators, could obligate us to stop using some of our clinical samples, which in turn may hinder our product development efforts. Such a result would also likely involve legal challenges that may consume our management and financial resources.
Failure to timely or accurately bill and collect for our services could have a material adverse effect on our revenues and our business.
Billing for laboratory testing services is extremely complicated and is subject to extensive and non-uniform rules and administrative requirements. Depending on the billing arrangement and applicable law, we bill various payors, such as patients, insurance companies, Medicare, Medicaid, physicians, hospitals and employer groups. Changes in laws and regulations and payor practices increase the complexity and cost of our billing process. Additionally, in the U.S., third-party payors generally require billing codes on claims for reimbursement that describe the services provided. For laboratory services, the American Medical Association establishes most of the billing codes using a data code set called Current Procedural Terminology, or CPT, codes and the World Health Organization establishes diagnostic codes using a data set called International Statistical Classification of Diseases, or ICD-10, codes. Each third-party payor generally develops payment amounts and coverage policies for their beneficiaries or members that ties to the CPT code established for the laboratory test and the ICD-10 code selected by the ordering or performing physician. Therefore, coverage and reimbursement may differ by payor even if the same billing code is reported for claims filing purposes. For laboratory tests without a specific billing code, payors often review claims on a claim-by-claim basis and there are increased uncertainties as to coverage and eligibility for reimbursement.
In addition to the items described above, third-party payors, including government programs, may decide to deny payment or recoup payments for testing that they contend was improperly billed or not medically necessary, against their coverage determinations, or for which they believe they have otherwise overpaid (including as a result of their own error), and we may be required to refund payments already received. Our revenues may be subject to retroactive adjustment as a result of these factors among others, including without limitation, differing interpretations of billing and coding guidance and changes by government agencies and payors in interpretations, requirements, and “conditions of participation” in various programs.
We have in the ordinary course of business been the subject of recoupments by payors and have from time to time identified and reimbursed payors for overpayments.
Incorrect or incomplete documentation and billing information, as well as the other items described above, among other factors, could result in non-payment for services rendered or having to pay back amounts incorrectly billed and collected. Further, the failure to timely or correctly bill could lead to various penalties, including: (1) exclusion from participation in the CMS and other government programs; (2) asset forfeitures; (3) civil and criminal fines and penalties; and (4) the loss of various licenses, certificates and authorizations necessary to operate our business, any of which could have a material adverse effect on our results of operations or cash flows.
38
The information technology systems that we rely on may be subject to unauthorized tampering, cyberattack or other data security incidents that could impact our billing processes or disrupt our operations
In addition to our internal information technology systems, we rely on the IT systems of certain third parties to whom we outsource certain of our services or functions, or with whom we store confidential information, including patient data. These IT systems are subject to potential cyberattacks or other security breaches. If such attacks are successful, they could disrupt our operations and result in unauthorized persons gaining access to confidential or proprietary information. A breach or security incident affecting these third parties could harm our business, results of operations and reputation, and subject us to liability, governmental investigation, significant damage to our reputation or otherwise adversely affect our business.
Although the Company has security measures implemented, cyber-attacks and threats against us and our third-party providers continue to evolve and are often not recognized until such attacks are launched against a potential target. A successful cybersecurity attack or other data security incident could result in the misappropriation and/or loss of confidential or personal information, create system interruptions, or deploy malicious software that attacks our systems. The unauthorized dissemination of sensitive personal information or proprietary or confidential information due to a breach of these IT systems could expose us or other third-parties to regulatory fines or penalties, litigation and potential liability, or otherwise harm our business. Any mitigation or remediation efforts that we undertake may require expenditures of significant resources and the diversion of the attention of management. In addition, we have taken, and continue to take, precautionary measures to reduce the risk of, and detect and respond to, future cyber threats, and prevent or minimize vulnerabilities in our IT systems. We have also taken, and will continue to take, measures to assess the cybersecurity protections implemented by our third-party providers. There can be no assurances that our precautionary measures or measures used by our third-party providers will prevent, contain or successfully defend against cyber or information security threats that could have a significant impact on our business, results of operations and reputation and subject us to liability.
Healthcare plans have taken steps to control the utilization and reimbursement of healthcare services, including clinical test services.
We also face efforts by non-governmental third-party payors, including healthcare plans, to reduce utilization and reimbursement for clinical testing services.
The healthcare industry has experienced a trend of consolidation among healthcare insurance plans, resulting in fewer but larger insurance plans with significant bargaining power to negotiate fee arrangements with healthcare providers, including clinical testing providers. These healthcare plans and independent physician associations, may demand that clinical testing providers accept discounted fee structures or assume all or a portion of the financial risk associated with providing testing services to their members through capped payment arrangements. In addition, some healthcare plans limit the laboratory network to only a single national or regional laboratory to obtain improved fee-for-service pricing. There is also an increasing number of patients enrolling in consumer driven products and high deductible plans that involve greater patient cost-sharing.
The increased consolidation among healthcare plans also has increased the potential adverse impact of ceasing to be a contracted provider with any such insurer.
We expect continuing efforts to limit the number of participating laboratories in payor networks, reduce reimbursements, to impose more stringent cost controls and to reduce utilization of clinical test services. These efforts, including future changes in third-party payor rules, practices and policies, or failing to become a contracted provider or ceasing to be a contracted provider to a healthcare plan, may have a material adverse effect on our business.
If we are unable to obtain and enforce patent protection for our products, our business could be materially harmed.
Our success depends, in part, on our ability to protect proprietary methods and technologies that we develop or license under the patent and other intellectual property laws of the U.S. and other countries, so that we can prevent others from unlawfully using our inventions and proprietary information. However, we may not hold proprietary rights to some patents required for us to commercialize our products and product candidates. Because certain U.S. patent applications are confidential, third parties may have filed patent applications for technology covered by our pending patent applications without our being aware of those applications, and our patent applications may not have priority over those applications. For this and other reasons, we or our third-party collaborators may be unable to secure desired patent rights, thereby losing desired exclusivity. If licenses are not available to us on acceptable terms, we may not be able to market the affected products or conduct the desired activities, unless we challenge the validity, enforceability, or infringement of the third-party patent or otherwise circumvent the third-party patent.
Our strategy depends on our ability to rapidly identify and seek patent protection for our discoveries. In addition, we will rely on third-party collaborators to file patent applications relating to proprietary technology that we develop jointly during
39
certain collaborations. The process of obtaining patent protection is expensive and time-consuming. If our present or future collaborators fail to file and prosecute all necessary and desirable patent applications at a reasonable cost and in a timely manner, our business will be adversely affected. Unauthorized parties may be able to obtain and use information that we regard as proprietary.
The issuance of a patent does not guarantee that it is valid or enforceable. Any patents we have obtained, or obtain in the future, may be challenged, invalidated, unenforceable, or circumvented. In addition, court decisions may introduce uncertainty in the enforceability or scope of patents owned by biotechnology, pharmaceutical, and medical device companies. Any challenge to, finding of unenforceability or invalidation or circumvention of, our patents or patent applications would be costly, would require significant time and attention of our management, and could have a material adverse effect on our business, results of operations and financial condition.
We cannot assure you that any patents that have issued, that may issue, or that may be licensed to us will be enforceable or valid, or will not expire prior to the commercialization of our products and product candidates, thus allowing others to more effectively compete with us. Therefore, any patents that we own or license may not adequately protect our products and product candidates or our future products, which could have a material adverse effect on our business, results of operations, and financial condition. We cannot be assured that our filings for patent term extensions or supplementary protection certificates to potentially extend a patent term of a patent covering an approved drug or biological product will be granted in any particular jurisdiction in which the Company or its licensee obtains approval for a drug or biological product.
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patent protection, we also rely on other proprietary rights, including protection of trade secrets, know-how, and confidential and proprietary information. To maintain the confidentiality of trade secrets and proprietary information, we will seek to enter into confidentiality agreements with our employees, consultants, and collaborators upon the commencement of their relationships with us. These agreements generally require that all confidential information developed by the individual or made known to the individual by us during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties. Our agreements with employees also generally provide that any inventions conceived by the individual in the course of rendering services to us shall be our exclusive property.
However, we may not obtain these agreements in all circumstances, and individuals with whom we have these agreements may not comply with their terms. In the event of unauthorized use or disclosure of our trade secrets or proprietary information, these agreements, even if obtained, may not provide meaningful protection, particularly for our trade secrets or other confidential information. To the extent that our employees, consultants, or contractors use technology or know-how owned by third parties in their work for us, disputes may arise between us and those third parties as to the rights in related inventions.
Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. The disclosure of our trade secrets would impair our competitive position and may materially harm our business, financial condition, and results of operations.
We will rely heavily on licenses from third parties. Failure to comply with the provisions of these licenses could result in the loss of our rights under the license agreements.
Many of the patents and patent applications in our patent portfolio are not owned by us, but are licensed from third parties. Such license agreements give us rights for the commercial exploitation of the patents resulting from the respective patent applications, subject to certain provisions of the license agreements. Failure to comply with these provisions could result in the loss of our rights under these license agreements. Our inability to rely on these patents and patent applications, which are the basis of our technology, would have a material adverse effect on our business, results of operations and financial condition.
We license patent rights to certain of our technology from third-party owners. If such owners do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects will be harmed.
We have obtained and may in the future obtain licenses from third party owners that are necessary or useful for our business. We cannot guarantee that no third parties will step forward and assert inventorship or ownership in our in-licensed patents. In some cases, we may rely on the assurances of our licensors that all ownership rights have been secured and that all necessary agreements are intact or forthcoming.
Our success will depend in part on our ability or the ability of our licensors to obtain, maintain, and enforce patent protection for our licensed intellectual property and, in particular, those patents to which we have secured exclusive rights in our field. We or our licensors may not successfully prosecute the patent applications which are licensed to us. Even if patents
40
issue in respect of these patent applications, we or our licensors may fail to maintain these patents or may determine not to pursue litigation against other companies that are infringing these patents. Without protection for the intellectual property we have licensed, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business, results of operations and financial condition.
Our commercial success depends significantly on our ability to operate without infringing the patents and other proprietary rights of third parties.
Other entities may have or obtain patents or proprietary rights that could limit our ability to develop, manufacture, use, sell, offer for sale or import products, or impair our competitive position. In addition, other entities may have or obtain patents or proprietary rights that cover our current research and preclinical studies. The U.S. case law pertaining to statutory exemptions to patent infringement for those who are using third party patented technology in the process of pursuing FDA regulatory approval changes over time. Lawsuits involving such exemptions are very fact intensive and it is currently unclear under U.S. case law whether preclinical studies would always qualify for such an exemption, and whether such exemptions would apply to research tools. To the extent that our current research and preclinical studies may be covered by the patent rights of others, the risk of suit may continue after such patents expire because the statute of limitations for patent infringement runs for six years. To the extent that a third party develops and patents technology that covers our products, we may be required to obtain licenses to that technology, which licenses may not be available or may not be available on commercially reasonable terms, if at all. If licenses are not available to us on acceptable terms, we will not be able to market the affected products or conduct the desired activities, unless we challenge the validity, enforceability or infringement of the third-party patent, or circumvent the third-party patent, which would be costly and would require significant time and attention of our management. Third parties may have or obtain by license or assignment valid and enforceable patents or proprietary rights that could block us from developing products using our technology. Our failure to obtain a license to any technology that we require may materially harm our business, financial condition, and results of operations.
If we become involved in patent litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, substantial liability for damages or be required to stop our product development and commercialization efforts.
Third parties may sue us for infringing their patent rights. Likewise, we may need to resort to litigation to enforce a patent issued or licensed to us or to determine the scope and validity of proprietary rights of others. In addition, a third-party may claim that we have improperly obtained or used its confidential or proprietary information. Furthermore, in connection with our third-party license agreements, we may have agreed to indemnify the licensor for costs incurred in connection with litigation relating to intellectual property rights. The cost to us of any litigation or other proceeding relating to intellectual property rights, even if resolved in our favor, could be substantial, and the litigation would divert our management’s efforts. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of any litigation could limit our ability to continue our operations. Our involvement in patent litigation and other proceedings could have a material adverse effect on our business, results of operations, and financial condition.
If any parties successfully claim that our creation or use of proprietary technologies infringes upon their intellectual property rights, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. In addition to any damages we might have to pay, a court could require us to stop the infringing activity or obtain a license. Any license required under any patent may not be made available on commercially acceptable terms, if at all. In addition, such licenses are likely to be non-exclusive and, therefore, our competitors may have access to the same technology licensed to us. If we fail to obtain a required license and are unable to design around a patent, we may be unable to effectively market some of our technology and products, which could limit our ability to generate revenues or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations.
We have faced, and may in the future face, intellectual property infringement claims that could be time-consuming and costly to defend, and could result in our loss of significant rights and the assessment of treble damages.
We may from time to time receive notices of claims of infringement and misappropriation or misuse of other parties’ proprietary rights. Some of these additional claims may also lead to litigation. We cannot assure you that we will prevail in such actions, or that other actions alleging misappropriation or misuse by us of third-party trade secrets, infringement by us of third-party patents and trademarks or the validity of our patents, will not be asserted or prosecuted against us.
We may also initiate claims to defend our intellectual property or to seek relief on allegations that we use, sell, or offer to sell technology that incorporates third party intellectual property. Intellectual property litigation, regardless of outcome, is expensive and time-consuming, could divert management’s attention from our business and have a material negative effect on our business, operating results or financial condition. If there is a successful claim of infringement against us, we may be
41
required to pay substantial damages (including treble damages if we were to be found to have willfully infringed a third party’s patent) to the party claiming infringement, develop non-infringing technology, stop selling our tests or using technology that contains the allegedly infringing intellectual property or enter into royalty or license agreements that may not be available on acceptable or commercially practical terms, if at all. Our failure to develop non-infringing technologies or license the proprietary rights on a timely basis could harm our business.
It is possible that in the patent laws related to the field of genomic-based products and diagnostics and patents covering such products changes to permit the patenting of genes and/or gene based products and/or related diagnostic methods. In such a case, we might be required to pay royalties, damages and costs to firms who own the rights to these patents, or we might be restricted from using any of the inventions claimed in those patents.
We may become subject to product liability for our diagnostic tests, clinical trials, pharmaceutical products and medical device products.
Our success depends on the market’s confidence that we can provide reliable, high-quality pharmaceuticals, medical devices, and diagnostics tests. Our reputation and the public image of our products or technologies may be impaired if our products fail to perform as expected or our products are perceived as difficult to use. Our products are complex and may develop or contain undetected defects or errors. Furthermore, if a product or future product candidate harms people, or is alleged to be harmful, we may be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, corporate partners or others. We have product liability insurance covering commercial sales of current products and our ongoing clinical trials. Any defects or errors could lead to the filing of product liability claims, which could be costly and time-consuming to defend and result in substantial damages. If we experience a sustained material defect or error, this could result in loss or delay of revenues, delayed market acceptance, damaged reputation, diversion of development resources, legal claims, increased insurance costs or increased service and warranty costs, any of which could materially harm our business. We cannot assure you that our product liability insurance would protect our assets from the financial impact of defending a product liability claim. A product liability claim could have a serious adverse effect on our business, financial condition and results of operations.
Adverse results in material litigation matters or governmental inquiries could have a material adverse effect upon our business and financial condition.
We may from time to time become subject in the ordinary course of business to material legal action related to, among other things, intellectual property disputes, professional liability, contractual and employee-related matters, as well as inquiries from governmental agencies and Medicare or Medicaid carriers requesting comment and information on allegations of billing irregularities and other matters that are brought to their attention through billing audits, third parties or other sources. The health care industry is subject to substantial federal and state government regulation and audit.
From time to time, we may receive inquiries, document requests, Civil Investigative Demands (“CIDs”) or subpoenas from the Department of Justice, the Office of Inspector General and Office for Civil Rights (“OCR”) of the Department of Health and Human Services, the Centers for Medicare and Medicaid Services, various payors and fiscal intermediaries, and other state and federal regulators regarding investigations, audits and reviews. We are currently responding to CIDs, subpoenas or document requests for various matters relating to our laboratory operations. Some pending or threatened proceedings against us may involve potentially substantial amounts as well as the possibility of civil, criminal, or administrative fines, penalties, or other sanctions, which could be material. Settlements of suits involving the types of issues that we routinely confront may require monetary payments as well as corporate integrity agreements. Additionally, qui tam or “whistleblower” actions initiated under the civil False Claims Act may be pending but placed under seal by the court to comply with the False Claims Act’s requirements for filing such suits. The Company generally has cooperated, and intends to continue to cooperate, with appropriate regulatory authorities as and when investigations, audits and inquiries arise.
Such legal actions and government investigations could result in substantial monetary damages, negatively impact our ability to obtain additional funding on acceptable terms, or at all, and damage to our reputation with customers, business partners and other third parties, all of which could have a material adverse effect upon our results of operations and financial position. Further, the legal actions and government investigations could damage our reputation with investors and adversely affect the trading prices of our securities.
RISKS RELATED TO REGULATORY COMPLIANCE
Our ability to successfully operate our laboratories and develop and commercialize certain of our diagnostic tests and LDTs will depend on our ability to maintain required regulatory licensures and comply with all the CLIA requirements.
42
In order to successfully operate our laboratory business and offer certain of our diagnostic tests and LDTs, we must maintain our CLIA certification and comply with all the CLIA requirements. CLIA is designed to ensure the quality and reliability of clinical laboratories by mandating specific standards in the areas of personnel qualifications, administration and participation in proficiency testing, patient test management, quality control, quality assurance and inspections. The sanction for failure to comply with CLIA requirements may be suspension, revocation or limitation of a laboratory’s CLIA certificate, which is necessary to conduct business, as well as significant fines and/or criminal penalties. Laboratories must undergo on-site surveys at least every two years, which may be conducted by the Federal CLIA program or by a private CMS approved accrediting agency such as CAP, among others. Our laboratories are also subject to regulation of laboratory operations under state clinical laboratory laws as will be any new CLIA-certified laboratory that we establish or acquire. State clinical laboratory laws may require that laboratories and/or laboratory personnel meet certain qualifications, specify certain quality controls or require maintenance of certain records. Certain states, such as California, Florida, Maryland, New York, Pennsylvania and Rhode Island, require that laboratories obtain licenses to test specimens from patients residing in those states and additional states may require similar licenses in the future. If we are unable to obtain and maintain licenses from states where required, we will not be able to process any samples from patients located in those states. Only Washington and New York States are exempt under CLIA, as these states have established laboratory quality standards at least as stringent as CLIA’s. Potential sanctions for violation of these statutes and regulations include significant fines and the suspension or loss of various licenses, certificates and authorizations, which could adversely affect our business and results of operations.
If we fail to comply with CLIA requirements, HHS or state agencies could require us to cease diagnostic testing. Even if it were possible for us to bring our laboratories back into compliance after failure to comply with such requirements, we could incur significant expenses and potentially lose revenues in doing so. Moreover, new interpretations of current regulations or future changes in regulations under CLIA may make it difficult or impossible for us to comply with the CLIA classification, which would significantly harm our business and materially adversely affect our financial condition.
The regulatory approval process is expensive, time consuming and uncertain and may prevent us or our collaboration partners from obtaining approvals for the commercialization of some or all of our product candidates.
The research, testing, manufacturing, labeling, approval, selling, marketing, and distribution of drug products, diagnostic products, or medical devices are subject to extensive regulation by the FDA and other non-U.S. regulatory authorities, which regulations differ from country to country. In general, we are not permitted to market our product candidates in the U.S. until we receive approval of a BLA, an approval of an NDA, a clearance letter under the premarket notification process, or 510(k) process, or an approval of a PMA from the FDA. To date, we have only submitted one NDA which was approved in June 2016, and one BLA which was approved for filing in January 2021. We have received FDA approval of the PMA for our Sangia Total PSA Test using the Claros Analyzer, a novel diagnostic instrument system to provide rapid, high performance blood test results in the point-of-care setting, in January 2019 and, in December 2021, for our 4Kscore test for use in men age 45 and older who have not had a prior prostate biopsy or are biopsy negative and have an age-specific abnormal total PSA and/or abnormal digital rectal exam, but we have not received marketing approval or clearance for any of our other diagnostic product candidates. In response to the BLA we submitted for Somatrogon Pfizer received a Complete Response Letter, which we and Pfizer are reviewing to determine the best path forward for Somatrogon. Obtaining approval of a NDA or PMA can be a lengthy, expensive, and uncertain process. With respect to medical devices, while the FDA reviews and clears a premarket notification in as little as three months, there is no guarantee that our products will qualify for this more expeditious regulatory process, which is reserved for Class I and II devices, nor is there any assurance that even if a device is reviewed under the 510(k) process that the FDA will review it expeditiously or determine that the device is substantially equivalent to a lawfully marketed non-PMA device. If the FDA fails to make this finding, then we cannot market the device. In lieu of acting on a premarket notification, the FDA may seek additional information or additional data which would further delay our ability to market the product. Furthermore, we are not permitted to make changes to a device approved through the PMA or 510(k) which affects the safety or efficacy of the device without first submitting a supplement application to the PMA and obtaining FDA approval or cleared premarket notification for that supplement. In some cases, the FDA may require clinical trials to support a supplement application. In addition, failure to comply with FDA, non-U.S. regulatory authorities, or other applicable U.S. and non-U.S. regulatory requirements may, either before or after product approval or clearance, if any, subject our company to administrative or judicially imposed sanctions, including, but not limited to the following:
▪restrictions on the products, manufacturers, or manufacturing process;
▪adverse inspectional observations (Form 483), warning letters, or non-warning letters incorporating inspectional observations;
▪civil and criminal penalties;
▪injunctions;
43
▪suspension or withdrawal of regulatory approvals or clearances;
▪product seizures, detentions, or import bans;
▪voluntary or mandatory product recalls and publicity requirements;
▪total or partial suspension of production;
▪imposition of restrictions on operations, including costly new manufacturing requirements; and
▪refusal to approve or clear pending NDAs or supplements to approved NDAs, applications or pre-market notifications.
Regulatory approval of an NDA or NDA supplement, BLA, PMA, PMA supplement or clearance pursuant to a pre-market notification is not guaranteed, and the approval or clearance process, as the case may be, is expensive and may, especially in the case of an NDA or PMA application, take several years. The FDA also has substantial discretion in the drug and medical device approval and clearance process. Failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials that will be required for FDA approval or clearance varies depending on the drug or medical device candidate, the disease or condition that the drug or medical device candidate is designed to address, and the regulations applicable to any particular drug or medical device candidate. The FDA can delay, limit or deny approval or clearance of a drug or medical device candidate for many reasons, including:
▪a drug candidate may not be deemed safe or effective;
▪a medical device candidate may not be deemed to be substantially equivalent to a lawfully marketed non-PMA device, in the case of a premarket notification;
▪the FDA may not find the data from pre-clinical studies and clinical trials sufficient;
▪the FDA may not approve our or our third-party manufacturer’s processes or facilities; or
▪the FDA may change its approval or clearance policies or adopt new regulations.
Beyond these risks, there is also a possibility that our licensees or collaborators could decide to discontinue a study at any time for commercial, scientific or other reasons.
The terms of approvals and ongoing regulation of our products may limit how we manufacture and market our products and product candidates, which could materially impair our ability to generate anticipated revenues.
We, our approved or cleared products, and the manufacturers of our products are subject to continual review. Our approved or cleared products may only be promoted for their indicated uses. Marketing, labeling, packaging, adverse event reporting, storage, advertising, and promotion for our approved products will be subject to extensive regulatory requirements. We train our marketing and sales force against promoting our products for uses outside of the cleared or approved indications for use, known as “off-label uses.” If the FDA determines that our promotional materials or training constitute promotion of unsupported claims or an off-label use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, injunction, seizure, civil fine or criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and/or administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, and the curtailment of our operations.
We and the manufacturers of our products are also required to comply with current Good Manufacturing Practices (“cGMP”) regulations or the FDA’s QSR regulations, which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Moreover, device manufacturers are required to report adverse events by filing Medical Device Reports with the FDA, which reports are publicly available.
Further, regulatory agencies must approve manufacturing facilities before they can be used to manufacture our products, and these facilities are subject to ongoing regulatory inspection. If we fail to comply with the regulatory requirements of the FDA and other non-U.S. regulatory authorities, or if previously unknown problems with our products, manufacturers, or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions. Furthermore, any limitation on indicated uses for a product or product candidate or our ability to manufacture and promote a product or product candidate could significantly and adversely affect our business, results of operations, and financial condition.
44
In addition, the FDA and other non-U.S. regulatory authorities may change their policies and additional regulations may be enacted that could prevent or delay marketing approval or clearance of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are not able to maintain regulatory compliance, we would likely not be permitted to market our products or product candidates and we may not achieve or sustain profitability, which would materially impair our ability to generate anticipated revenues.
If we fail to comply with complex and rapidly evolving laws and regulations, we could suffer penalties, be required to pay substantial damages or make significant changes to our operations.
We are subject to numerous federal and state regulations, including, but not limited to:
•federal and state laws applicable to billing and claims payment;
•federal and state laboratory anti-mark-up laws;
•federal and state anti-kickback laws;
•physician self-referral law;
•federal and state false claims laws;
•federal self-referral and financial inducement prohibition laws, commonly known as the Stark Law, and the state equivalents;
•federal and state laws governing laboratory licensing and testing, including CLIA;
•federal and state laws governing the development, use and distribution of LDTs;
•HIPAA, along with the revisions to HIPAA as a result of the amendments from the Health Information Technology for Economic and Clinical Health Act of 2009 (“HITECH Act”), and analogous state laws and non-US laws, including the General Data Protection Regulation;
•federal, state and foreign regulation of privacy, security, electronic transactions and identity theft;
•federal, state and local laws governing the handling, transportation and disposal of medical and hazardous waste;
•Occupational Safety and Health Administration rules and regulations;
•changes to laws, regulations and rules as a result of the implementation and/or repeal of part or all of 2010 Health Care Reform Legislation; and
•changes to other federal, state and local laws, regulations and rules, including tax laws.
If we fail to comply with existing or future applicable laws and regulations, we could suffer civil or criminal penalties, including the loss of our licenses to operate our laboratories and our ability to participate in federal and state healthcare programs. Different interpretations and enforcement policies of existing statutes and regulations applicable to our business could subject our current practices to allegations of impropriety or illegality, or could require us to make significant changes to our operations. Under the False Claims Act (“FCA”), whistleblower or qui tam provisions allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought by private individuals has increased dramatically and we may be subject to such suits. Violations of the FCA could result in enormous economic liability and could have a material impact on us. As a result of political, economic, and regulatory influences, the healthcare delivery industry in the U.S. is under intense scrutiny and subject to fundamental changes. We cannot predict which reform proposals will be adopted, when they may be adopted, or what impact they may have on us. The costs associated with complying with federal and state regulations could be significant and the failure to comply with any such legal requirements could have a material adverse effect on our financial condition, results of operations, and liquidity.
45
Failure to maintain the security of patient-related information or compliance with security requirements could damage our reputation with customers, cause us to incur substantial additional costs and become subject to litigation.
Pursuant to HIPAA, including the HITECH amendments thereunder, and certain similar state laws, we must comply with comprehensive privacy and security standards with respect to the use and disclosure of protected health information. If we do not comply with existing or new laws and regulations related to protecting privacy and security of personal or health information, we could be subject to monetary fines, civil penalties, or criminal sanctions.
We may also be required to comply with the data privacy and security laws of other countries in which we operate or from which we receive data transfers, including the General Data Protection Regulation (GDPR), which affects our European operations and possibly our laboratory and clinical development operations. The GDPR, which is wide-ranging in scope, governs the collection and use of personal data in the European Union and imposes operational requirements for companies that receive or process personal data of residents of the European Union that are different than those currently in place in the European Union. We have implemented policies and procedures required to comply with the new EU regulations but may be subject for penalties if we are found to be non-compliant.
We have had data and security breaches in the ordinary course and such breaches may continue to happen from time to time despite our best efforts to prevent such breaches and safeguard private information. Some of these other data and security breaches have been reported to OCR and we have received requests for information from OCR in connection with certain of these matters, or we are awaiting discussion, investigation or action by OCR. Any action by OCR may require us to pay fines or take remedial actions that may be expensive and require the attention of management, any of which may have a material adverse effect on us and our results of operations.
We have and will continue to receive certain personal and financial information about our clients and their patients. In addition, we depend upon the secure transmission of confidential information over public networks. While we take reasonable and prudent steps to protect this protected information, a compromise in our security systems that results in client or patient personal information being obtained by unauthorized persons or our failure to comply with security requirements for financial transactions could adversely affect our reputation with our clients and result in litigation against us or the imposition of penalties, all of which may adversely impact our results of operations, financial condition and liquidity.
Failure to comply with environmental, health and safety laws and regulations, including the Federal Occupational Safety and Health Administration Act, the Needlestick Safety and Prevention Act and the Comprehensive Medical Waste Management Act, could result in fines and penalties and loss of licensure, and have a material adverse effect upon our business.
We are subject to licensing and regulation under federal, state and local laws and regulations relating to the protection of the environment and human health and safety, including laws and regulations relating to the handling, transportation and disposal of medical specimens, infectious and hazardous waste and radioactive materials, as well as regulations relating to the safety and health of laboratory employees. The Federal Occupational Safety and Health Administration has established extensive requirements relating to workplace safety for health care employers, including clinical laboratories, whose workers may be exposed to blood-borne pathogens such as HIV and the hepatitis B virus. These requirements are designed to minimize exposure to, and transmission of, blood-borne pathogens. In addition, the Needlestick Safety and Prevention Act requires, among other things, that we include in our safety programs the evaluation and use of engineering controls such as safety needles if found to be effective at reducing the risk of needlestick injuries in the workplace.
Waste management is subject to federal and state regulations governing the transportation and disposal of medical waste including bodily fluids. In New Jersey, we are subject to the Comprehensive Medical Waste Management Act (“CMWMA”), which requires us to register as a generator of special medical waste. All of our medical waste is disposed of by a licensed interstate hauler. These records are audited by the State of New Jersey on a yearly basis. We are also subject to the Federal Hazardous Materials Transportation Law, 49 U.S.C. 5101 et seq., and the Hazardous Materials Regulations (“HMR”), 49 CFR parts 171-180. The federal government has classified hazardous medical waste as hazardous materials for the purpose of regulation.
Failure to comply with such federal, state and local laws and regulations could subject us to denial of the right to conduct business, fines, criminal penalties and/or other enforcement actions, any of which could have a material adverse effect on our business. In addition, compliance with future legislation could impose additional requirements us, which may be costly.
46
Our failure or the failure of third-party payors or physicians to comply with ICD-10-CM Code Set, and our failure to comply with other emerging electronic transaction standards could adversely impact our business.
We continue our assessment of information systems, applications and processes for compliance with ICD-10-CM Code Set requirements. Clinical laboratories are typically required to submit health care claims with diagnosis codes to third party payors. The diagnosis codes must be obtained from the ordering physician for clinical laboratory testing and from the interpreting pathologist for anatomic pathology services. Our failure or the failure of third party payors or physicians to comply with these requirements could have an adverse impact on reimbursement, delay sales and cash collections.
Also, the failure of our IT systems to keep pace with technological advances may significantly reduce our revenues or increase our expenses. Public and private initiatives to create healthcare information technology (“HCIT”) standards and to mandate standardized clinical coding systems for the electronic exchange of clinical information, including test orders and test results, could require costly modifications to our existing HCIT systems. If we fail to adopt or delay in implementing HCIT standards, we could lose customers and business opportunities.
Failure to comply with complex federal and state laws and regulations related to submission of claims for clinical laboratory services could result in significant monetary damages and penalties and exclusion from the Medicare and Medicaid programs.
We are subject to extensive federal and state laws and regulations relating to the submission of claims for payment for clinical laboratory services, including those that relate to coverage of our services under Medicare, Medicaid and other governmental health care programs, the amounts that may be billed for our services and to whom claims for services may be submitted. These rules may also affect us in light of the practice management products that we market, to the extent that these products are considered to affect the manner in which our customers submit their own claims for services. Submission of our claims is particularly complex because we provide both anatomic pathology services and clinical laboratory tests, which generally are paid using different reimbursement principles. The clinical laboratory tests are often paid under a clinical laboratory fee schedule, and the anatomic pathology services are often paid under a physician fee schedule.
Our failure to comply with applicable laws and regulations could result in our inability to receive payment for our services or result in attempts by third-party payors, such as Medicare and Medicaid, to recover payments from us that have already been made. Submission of claims in violation of certain statutory or regulatory requirements can result in penalties, including substantial civil money penalties for each item or service billed to Medicare in violation of the legal requirement, and exclusion from participation in Medicare and Medicaid. Government authorities may also assert that violations of laws and regulations related to submission or causing the submission of claims violate the FCA or other laws related to fraud and abuse, including submission of claims for services that were not medically necessary. Under the FCA, whistleblower or qui tam provisions allow a private individual to bring actions on behalf of the federal government alleging that the defendant has submitted a false claim to the federal government, and to share in any monetary recovery. In recent years, the number of suits brought by private individuals has increased dramatically and we may be subject to such suits. Violations of the FCA could result in enormous economic liability. The FCA provides that all damages are trebled, and each false claim submitted is subject to a penalty of up to $21,916. For example, we could be subject to FCA liability if it was determined that the services we provided were not medically necessary and not reimbursable, particularly if it were asserted that we contributed to the physician’s referrals of unnecessary services to us. It is also possible that the government could attempt to hold us liable under fraud and abuse laws for improper claims submitted by an entity for services that we performed if we were found to have knowingly participated in the arrangement that resulted in submission of the improper claims.
Changes in regulation and policies, including increasing downward pressure on health care reimbursement, may adversely affect reimbursement for diagnostic services and could have a material adverse impact on our business.
Reimbursement levels for health care services are subject to continuous and often unexpected changes in policies, and we face a variety of efforts by government payors to reduce utilization and reimbursement for diagnostic testing services. Changes in governmental reimbursement may result from statutory and regulatory changes, retroactive rate adjustments, administrative rulings, competitive bidding initiatives, and other policy changes.
The U.S. Congress has considered, at least yearly in conjunction with budgetary legislation, changes to one or both of the Medicare fee schedules under which we receive reimbursement, which include the physician fee schedule for anatomical pathology services, and the clinical laboratory fee schedule for our clinical laboratory services. For example, currently there is no copayment or coinsurance required for clinical laboratory services, although there is for our services that are paid under the physician fee schedule. However, Congress has periodically considered imposing a 20 percent coinsurance on laboratory services. If enacted, this would require us to attempt to collect this amount from patients, although in many cases the costs of collection would exceed the amount actually received.
47
The Center for Medicare and Medicaid Services (“CMS”) pays laboratories on the basis of a fee schedule that is reviewed and re-calculated on an annual basis. CMS may change the fee schedule upward or downward on billing codes that we submit for reimbursement on a regular basis. Our revenue and business may be adversely affected if the reimbursement rates associated with such codes are reduced. Even when reimbursement rates are not reduced, policy changes add to our costs by increasing the complexity and volume of administrative requirements. Medicaid reimbursement, which varies by state, is also subject to administrative and billing requirements and budget pressures. In recent years, state budget pressures have caused states to consider several policy changes that may impact our financial condition and results of operations, such as delaying payments, reducing reimbursement, restricting coverage eligibility and service coverage, and imposing taxes on our services.
Third party payors are increasingly challenging established prices, and new products that are more expensive than existing treatments may have difficulty finding ready acceptance unless there is a clear therapeutic benefit. On April 1, 2014, the PAMA was enacted into law. Under PAMA, Medicare payment for clinical diagnostic laboratory tests is established by calculating a weighted mean of private payor rates. Effective January 1, 2018, clinical laboratory fee schedule rates are based on weighted median private payor rates as required by PAMA. Even though the permitted annual decrease are capped through 2023, the cap does not apply to new tests or new advanced diagnostic tests. We cannot assure you that any of our products will be considered cost effective, or that reimbursement will be available or sufficient to allow us to sell them competitively and profitably.
The federal government is faced with significant economic decisions in the coming years. Some solutions being offered in the government could substantially change the way laboratory testing is reimbursed by government entities. We cannot be certain what or how any such government changes may affect our business.
Medicare legislation and future legislative or regulatory reform of the health care system may affect our ability to sell our products profitably.
In the U.S., there have been a number of legislative and regulatory initiatives, at both the federal and state government levels, to change the healthcare system in ways that, if approved, could affect our ability to sell our products and provide our laboratory services profitably. As such, we cannot assure you that reimbursement payments under governmental and private third party payor programs will remain at levels comparable to present levels or will be sufficient to cover the costs allocable to patients eligible for reimbursement under these programs. Any changes that lower reimbursement rates under Medicare, Medicaid or private payor programs could negatively affect our business.
Most significantly, on March 23, 2010, President Obama signed into law both the Affordable Care Act and the reconciliation law known as Health Care and Education Affordability Reconciliation Act (the “Reconciliation Act”) and, combined we refer to both Acts as the “2010 Health Care Reform Legislation.” The constitutionality of the 2010 Health Care Reform Legislation was confirmed on June 28, 2012 by the Supreme Court of the U. S.
It is uncertain whether any efforts to amend the Affordable Care Act will be successful or enacted into law, and if enacted, what the impact might be on our business. It is also uncertain how the current administration intends to alter 2010 Health Care Reform Legislation, if at all including whether regulatory changes to the implementation of the 2010 Health Care Reform Legislation will restrict patient access to affordable insurance or other third-party payor sources and impact their access to novel, biosimilar and complex generic products. In addition, litigation may prevent some or all of the legislation from taking effect. We cannot assure you as to the ultimate content, timing, or effect of changes, nor is it possible at this time to estimate the impact of any such potential legislation.
To enhance compliance with applicable health care laws, and mitigate potential liability in the event of noncompliance, regulatory authorities, such as the U. S. Health and Human Services Department Office of Inspector General (the “OIG”), have recommended the adoption and implementation of a comprehensive health care compliance program that generally contains the elements of an effective compliance and ethics program. In addition, certain states, such as New York, require that certain health care providers have a compliance program that generally adheres to the standards set forth in a model compliance program. Also, under the 2010 Health Care Reform Legislation, the U.S. Department of Health and Human Services, or HHS, requires suppliers, such as us, to adopt, as a condition of Medicare participation, compliance programs that meet a core set of requirements. While we have adopted U.S. healthcare compliance and ethics programs that generally incorporate the OIG’s recommendations and train our employees in such compliance, having such a program can be no assurance that we will avoid any compliance issues.
RISKS RELATED TO INTERNATIONAL OPERATIONS
Failure to obtain regulatory approval outside the U.S. will prevent us from marketing our products and product candidates abroad.
48
We intend to market certain of our products and product candidates in non-U.S. markets. In order to market our products and product candidates in the European Union and many other non-U.S. jurisdictions, we must obtain separate regulatory approvals. We have had limited interactions with non-U.S. regulatory authorities, the approval procedures vary among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval or clearance. Approval or clearance by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more non-U.S. regulatory authority does not ensure approval by other regulatory authorities in other countries or by the FDA. The non-U.S. regulatory approval process may include all of the risks associated with obtaining FDA approval or clearance. We may not obtain non-U.S. regulatory approvals on a timely basis, if at all. We may not be able to file for non-U.S. regulatory approvals and may not receive necessary approvals to commercialize our products and product candidates in any market, which would have a material adverse effect on our business, results of operations and financial condition.
Non-U.S. governments often impose strict price controls, which may adversely affect our future profitability.
We intend to seek approval to market certain of our products and product candidates in both the U.S. and in non‑U.S. jurisdictions. If we obtain approval in one or more non-U.S. jurisdictions, we will be subject to rules and regulations in those jurisdictions relating to our product. In some countries, particularly countries of the European Union, each of which has developed its own rules and regulations, pricing is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a drug or medical device candidate. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product and product candidates to other available products. If reimbursement of our products and product candidates is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, we may be unable to generate revenues and achieve or sustain profitability, which would have a material adverse effect on our business, results of operations and financial condition.
Potential political, economic and military instability in the State of Israel, where we have office, laboratory and manufacturing operations, may adversely affect our results of operations.
We maintain office, laboratory and manufacturing facilities in the State of Israel. Political, economic and military conditions in Israel may directly affect our ability to conduct business. Since the State of Israel was established in 1948, a number of armed conflicts have occurred between Israel and its neighbors. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners, or a significant downturn in the economic or financial condition of Israel, could affect adversely our operations. Ongoing and revived hostilities or other Israeli political or economic factors could harm our operations and product development and cause our revenues to decrease.
Due to the international scope of our business activities, our results of operations may be significantly affected by currency fluctuations.
We derive a significant portion of our consolidated net revenues from international sales, subjecting us to risks relating to fluctuations in currency exchange rates. Currency variations can adversely affect margins on sales of our products in countries outside of the U.S. and margins on sales of products that include components obtained from suppliers located outside of the U.S. Through our subsidiaries, we operate in a wide variety of jurisdictions. Certain countries in which we operate or may operate have experienced geopolitical instability, economic problems and other uncertainties from time to time. To the extent that world events or economic conditions negatively affect our future sales to customers in these and other regions of the world, or the collectability of receivables, our future results of operations, liquidity and financial condition may be adversely affected. We may manage exposures arising in the normal course of business related to fluctuations in foreign currency exchange rates by entering into offsetting positions through the use of foreign exchange forward contracts. Certain firmly committed transactions are hedged with foreign exchange forward contracts whereby exchange rates change, gains and losses on the exposed transactions are partially offset by gains and losses related to the hedging contracts. However, our subsidiaries receive their income and pay their expenses primarily in their local currencies. To the extent that transactions of these subsidiaries are settled in their local currencies, a devaluation of those currencies versus the U.S. dollar could reduce the contribution from these subsidiaries to our consolidated results of operations as reported in U.S. dollars. For financial reporting purposes, such depreciation will negatively affect our reported results of operations since earnings denominated in foreign currencies would be converted to U.S. dollars at a decreased value. While we have employed economic cash flow and fair value hedges to minimize the risks associated with these exchange rate fluctuations, the hedging activities may be ineffective or may not offset more than a portion of the adverse financial impact resulting from currency variations. Accordingly, we cannot assure you that fluctuations in the values of the currencies of countries in which we operate will not materially adversely affect our future results of operations.
49
We may be exposed to liabilities under the Foreign Corrupt Practices Act, and any determination that we violated the Foreign Corrupt Practices Act could have a material adverse effect on our business.
We are subject to the FCPA and other laws that prohibit U.S. companies or their agents and employees from providing anything of value to a foreign official or political party for the purposes of influencing any act or decision of these individuals in their official capacity to help obtain or retain business, direct business to any person or corporate entity or obtain any unfair advantage. We have operations and agreements with third parties and we generate sales internationally. Our international activities create the risk of unauthorized and illegal payments or offers of payments by our employees, consultants, sales agents or distributors, even though they may not always be subject to our control. We discourage these practices by our employees and agents. However, our existing safeguards and any future improvements may prove to be less than effective, and our employees, consultants, sales agents or distributors may engage in conduct for which we might be held responsible. Any failure by us to adopt appropriate compliance procedures and ensure that our employees and agents comply with the FCPA and applicable laws and regulations in foreign jurisdictions could result in substantial penalties or restrictions on our ability to conduct business in certain foreign jurisdictions.
Violations of the FCPA may result in severe criminal or civil sanctions, and we may be subject to other liabilities, which could negatively affect our business, operating results and financial condition. In addition, the U.S. government may seek to hold our Company liable for successor liability FCPA violations committed by companies in which we invest or that we acquire.
We are subject to risks associated with doing business globally.
Our operations, both within and outside the U.S., are subject to risks inherent in conducting business globally and under the laws, regulations and customs of various jurisdictions and geographies. These risks differ in some respects from those associated with our U.S. business and our exposure to such risks may increase if our international business continues to grow. These risks include fluctuations in currency exchange rates, changes in exchange controls, loss of business in government tenders that are held annually in many cases, nationalization, increasingly complex labor environments, expropriation and other governmental actions, changes in taxation, including legislative changes in U.S. and international taxation of income earned outside of the U.S., importation limitations, export control restrictions, violations of U.S. or local laws, including the FCPA, dependence on a few government entities as customers, pricing restrictions, economic destabilization, political and economic instability and disruption or destruction in a significant geographic region - due to the location of manufacturing facilities, distribution facilities or customers - regardless of cause, including war, terrorism, riot, civil insurrection or social unrest, or natural or man-made disasters, including famine, flood, fire, earthquake, storm or disease.
Our international business is subject to both U.S. and foreign laws and regulations, including, without limitation, regulations relating to import-export controls, technology transfer restrictions, repatriation of earnings, data privacy and protection, investment, exchange rates and controls, the FCPA and other anti-corruption laws, the anti-boycott provisions of the U.S. Export Administration Act, labor and employment, works councils and other labor groups, taxes, environment, security restrictions, intellectual property, changes in taxation, including legislative changes in U.S. and international taxation of income earned outside of the U.S., handling of regulated substances, and other commercial activities. Failure by us, our employees, affiliates, partners or others with whom we work to comply with these laws and regulations could result in administrative, civil or criminal liabilities. New regulations and requirements, or changes to existing ones in the various countries in which we operate can significantly increase our costs and risks of doing business internationally. Failure to comply with the laws and regulations that affect our global operations, could have an adverse effect on our business, financial condition or results of operations.
Changes in regulations, political leadership and environment, or security risks may dramatically affect our ability to conduct or continue to conduct business in international markets. Our international business may also be impacted by changes in foreign national policies and priorities, which may be influenced by changes in the environment, geopolitical uncertainties, government budgets, and economic and political factors more generally, any of which could impact funding for programs or delay purchasing decisions or customer payments. The occurrence and impact of these factors is difficult to predict, but one or more of them could have a material adverse effect on our financial position, results of operations and/or cash flows.
RISKS RELATED TO ACQUISITIONS AND INVESTMENTS
We have a large amount of goodwill and other intangible assets on our balance sheet that are subject to periodic impairment evaluations.
We have a large amount of goodwill and other intangible assets and we are required to perform an annual, or in certain situations a more frequent, assessment for possible impairment for accounting purposes. At December 31, 2021, we have goodwill and other intangible assets of $1.4 billion. Goodwill is tested at least annually for impairment or when events or
50
changes in circumstances indicate that the carrying amount of such assets may not be recoverable, by assessing qualitative factors or performing a quantitative analysis in determining whether it is more likely than not that its fair value exceeds the carrying value. Examples of qualitative factors include our share price, our financial performance compared to budgets, long-term financial plans, macroeconomic, industry and market conditions as well as the substantial excess of fair value over the carrying value of net assets from the annual impairment test previously performed.
Sales of Rayaldee and our operations at EirGen, are currently underperforming expectations and if we do not achieve our planned operating results, we may be required to incur a non-cash impairment charge. There can be no assurance that future reviews of our goodwill and other intangible assets will not result in impairment charges. Any impairment charges in the future will adversely affect our results of operations. A significant write down of goodwill and/or other intangible assets would have a material adverse effect on our reported results of operations and net worth and the trading price of our securities.
We submitted the initial BLA with the FDA for approval of Somatrogon (hGH-CTP) in the U.S. and Pfizer received a Complete Response Letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon in the United States. If we are unable to successfully commercialize Somatrogon in the U.S., or changes in projections and assumptions negatively impact our forecast of net cash flows, we may be exposed to a material impairment charge related to the IPR&D for Somatrogon.
RISKS RELATED TO OWNERSHIP OF OUR COMMON STOCK
The trading prices of our securities may fluctuate significantly.
The trading prices of our Common Stock may fluctuate significantly in response to numerous factors, some of which are beyond our control, such as:
•the announcement of new products or product enhancements by us or our competitors;
•results of our clinical trials and other development efforts;
•developments concerning intellectual property rights and regulatory approvals;
•variations in our and our competitors’ results of operations;
•changes in earnings estimates or recommendations by securities analysts, if our Common Stock is covered by analysts;
•developments in the biotechnology, pharmaceutical, diagnostic and medical device industry;
•the announcement and/or commencement and/or settlement of lawsuits or similar claims against us or any of our officers, directors and affiliates;
•the results of product liability or intellectual property lawsuits;
•future issuances of our Common Stock or other securities, including debt;
•purchases and sales of our Common Stock by our officers, directors or affiliates;
•the addition or departure of key personnel;
•announcements by us or our competitors of acquisitions, investments or strategic alliances; and
•general market conditions and other factors, including factors unrelated to our operating performance.
Further, the securities market in general, and the market for biotechnology, pharmaceutical, diagnostic and medical device companies in particular, has experienced extreme price and volume fluctuations in recent years. Continued market fluctuations could result in extreme volatility in the trading prices of our Common Stock, which could cause a decline in the value of our securities.
Directors, executive officers, principal stockholders and affiliated entities own a substantial amount of our capital stock, and they may make decisions that you do not consider to be in the best interests of our stockholders.
As of February 15, 2022, our directors, executive officers, principal stockholders and affiliated entities beneficially owned, in the aggregate, approximately 40% of our outstanding voting securities. Phillip Frost, M.D., our Chairman and CEO, is deemed to beneficially own, in the aggregate, approximately 33.9% of our Common Stock as of February 15, 2022. As a result, Dr. Frost, acting with other members of management, would have the ability to significantly impact the election of our Board of Directors, the adoption or amendment of provisions in our Certificate of Incorporation, the approval of mergers and other significant corporate transactions and the outcome of issues requiring approval by our stockholders. This concentration of
51
ownership may also have the effect of delaying or preventing a change in control of our company that may be favored by other stockholders. This could prevent transactions in which holders of our securities might otherwise recover a premium for their securities over current market prices.
A significant short position in our stock could have a substantial impact on the trading price of our stock.
Historically, there has been a significant “short” position in our Common Stock. As of January 31, 2022, investors held a short position of approximately 43,130,061 million shares of our Common Stock which represented approximately 6.3% of our outstanding Common Stock. The anticipated downward pressure on our stock price due to actual or anticipated sales of our stock by some institutions or individuals who engage in short sales of our Common Stock could cause our stock price to decline. Such stock price decrease could encourage further short-sales that could place additional downward pressure on our stock price. This could lead to further increases in the already large short position in our Common Stock and cause volatility in our stock price.
The volatility of our stock may cause the value of a stockholder’s investment to decline rapidly. Additionally, if our stock price declines, it may be more difficult for us to raise capital and may have other adverse effects on our business.
Failure to maintain effective internal controls in accordance with Section 404 of the Sarbanes-Oxley Act, including with respect to companies we acquire, could have a material adverse effect on our business and operating results. In addition, current and potential stockholders could lose confidence in our financial reporting, which could have a material adverse effect on the price of our Common Stock.
Section 404 of the Sarbanes-Oxley Act of 2002 requires annual management assessments of the effectiveness of our internal control over financial reporting and a report by our independent registered public accounting firm on the effectiveness of internal control over financial reporting as of year-end. We are required to report, among other things, control deficiencies that constitute material weaknesses or changes in internal control that, or that are reasonably likely to, materially affect internal control over financial reporting. A “material weakness” is a significant deficiency or combination of significant deficiencies that results in more than a remote likelihood that a material misstatement of the annual or interim financial statements will not be prevented or detected.
We have identified and remediated control deficiencies in the past, and we cannot assure you that we will at all times in the future be able to report that our internal controls are effective. In addition, material weaknesses in the design and operation of the internal control over financial reporting of companies that we acquire could have a material adverse effect on our business and operating results. If we cannot provide reliable financial reports or prevent fraud, our results of operation could be harmed. Our failure to maintain the effective internal control over financial reporting could cause the cost related to remediation to increase and could cause our stock price to decline. In addition, we may not be able to accurately report our financial results, may be subject to regulatory sanction, and investors may lose confidence in our financial statements.
Compliance with changing regulations concerning corporate governance and public disclosure may result in additional expenses.
There have been changing laws, regulations, and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, the Dodd-Frank Act, regulations promulgated by the Securities and Exchange Commission and rules promulgated by the Nasdaq Global Select Market and the other national securities exchanges. These new or changed laws, regulations, and standards are subject to varying interpretations in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. As a result, our efforts to comply with evolving laws, regulations, and standards are likely to continue to result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. Our board members, Chief Executive Officer, Chief Financial Officer, and Principal Accounting Officer could face an increased risk of personal liability in connection with the performance of their duties. As a result, we may have difficulty attracting and retaining qualified board members and executive officers, which could harm our business. If our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies, we could be subject to liability under applicable laws or our reputation may be harmed, which could materially adversely affect our business, results of operations and financial condition.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
52
ITEM 2. PROPERTIES.
Our principal corporate office is located at 4400 Biscayne Blvd, Miami, Florida. We lease this space from Frost Real Estate Holdings, LLC (“Frost Real Estate”), an entity which is controlled by Dr. Phillip Frost, our Chairman of the Board and Chief Executive Officer. Pursuant to the lease agreement with Frost Real Estate, we lease approximately 29,500 square feet, which encompasses space for our corporate offices and administrative services.
The table below summarizes certain information as to our significant physical properties as of December 31, 2021:
| Location | Segment and Purpose | Type of Occupancy | ||||||||||||
| Miami, FL | Diagnostics & Pharmaceutical: Corporate Headquarters | Leased | ||||||||||||
| Elmwood Park, NJ | Diagnostics: Main Laboratory | Leased | ||||||||||||
| Gaithersburg, MD | Diagnostics: Genetics Laboratory | Leased | ||||||||||||
| Kiryat Gat, Israel | Pharmaceutical: Research and Development, CTP | Leased | ||||||||||||
| Woburn, MA | Diagnostics | Leased | ||||||||||||
| Nesher, Israel | Pharmaceuticals: API Manufacturing | Leased | ||||||||||||
| Guadalajara, Mexico | Pharmaceuticals: Pharmaceutical Manufacturing | Owned | ||||||||||||
| Banyoles, Spain | Pharmaceuticals: Pharmaceutical Manufacturing | Owned | ||||||||||||
| Palol de Revardit, Spain | Warehouse | Leased | ||||||||||||
| Barcelona, Spain | Pharmaceuticals: Research and Development | Leased | ||||||||||||
| Waterford, Ireland | Pharmaceuticals: Pharmaceutical Manufacturing | Leased | ||||||||||||
| Santiago, Chile | Pharmaceuticals: Office; Warehouse | Leased | ||||||||||||
ITEM 3. LEGAL PROCEEDINGS.
We are involved from time to time in various claims and legal actions arising in the ordinary course of business.
As previously disclosed, on September 7, 2018, the SEC filed a lawsuit in the Southern District of New York (the “SEC Complaint” or “SEC Lawsuit”) against a number of individuals and entities, including the Company and its CEO and Chairman, Phillip Frost (“Dr. Frost”), alleging violations of securities laws. The Company entered into a settlement agreement with the SEC on December 27, 2018, pursuant to which, without admitting or denying any of the allegations of the SEC Complaint, the Company is enjoined from violating Section 13(d) of the Exchange Act and paid a $100,000 penalty.
Following the SEC’s announcement of the SEC Complaint, a number of class actions and derivative suits were filed concerning the allegations in the SEC Complaint and related matters. On June 26, 2020, The Amitim Funds, the lead plaintiff in the class action lawsuits filed a Stipulation of Settlement in the Southern District of Florida of behalf of itself and the remainder of the class, which provided for the settlement of and release of the class action claims for an aggregate of $16.5 million, a significant portion of which was paid into escrow by our insurance carriers. The settlement was approved on December 15, 2020. Additional information on the class action lawsuits is provided below.
On November 2, 2020, the derivative suit was settled in the Delaware Chancery Court for an aggregate amount of $3.1 million (the “Settlement”). Several derivative lawsuits that were filed in Florida State and federal courts were stayed pending the resolution of the Delaware derivative action and all of those matters have now been dismissed. Specific information relating to each lawsuit was previously disclosed and is provided below.
On or about September 12, 2018, Jason Kerznowski (“Kerznowski”), a purported stockholder, filed a putative class action lawsuit in the United States District Court for the District of New Jersey against the Company and certain of its current and former executive officers (the “Kerznowski Lawsuit”). This lawsuit was brought by Kerznowski both individually and on behalf of a putative class of the Company’s stockholders, claiming that in connection with the facts and circumstances underlying the allegations in the SEC Complaint, the Company engaged in fraudulent conduct and made false and misleading statements of material fact or omitted to state material facts necessary to make the statements made not misleading. The Kerznowski Lawsuit sought to declare the action to be a class action and certify Kerznowski as the class representative, monetary damages, an award of reasonable attorneys’ fees, expert fees, and other costs, and such other relief as the Court may deem just and proper.
53
On or about September 14, 2018, Charles Steinberg (“Steinberg”), a purported stockholder, filed a putative class action lawsuit in the United States District Court for the Southern District of Florida against the Company and certain of its current and former executive officers (the “Steinberg Lawsuit”). This lawsuit was brought by Steinberg both individually and on behalf of a putative class of the Company’s stockholders claiming that in connection with the facts and circumstances underlying the allegations in the SEC Complaint, the Company engaged in fraudulent conduct and made false and misleading statements of material fact or omitted to state material facts necessary to make the statements made not misleading. The Steinberg Lawsuit sought to declare the action to be a class action, monetary damages, an award of reasonable attorneys’ fees and expert fees and other costs, and such additional or different relief as the interests of law or equity may require.
On February 4, 2019, the United States District Court for the District of New Jersey appointed the Amitim Funds as lead plaintiff in the Kerznowski Lawsuit and subsequently transferred the matter to the United States District Court for the Southern District of Florida. Amitim Funds was also appointed as lead plaintiff in the Steinberg Lawsuit. On May 3, 2019, plaintiffs in each of the Kerznowski Lawsuit and Steinberg Lawsuit, filed an identical consolidated class action complaint, and the Court consolidated these actions on May 8, 2019. The class action was settled on December 15, 2020 for $16.5 million.
On or about September 13, 2018, Idan Sharon filed an Application for Approval of a Class Action in the Tel Aviv Israel District Court against the Company and certain of its current and former executive officers, and certain members of its Board of Directors (the “Sharon Claim”). This application was filed by a purported stockholder, both individually and on behalf of a putative class of the Company’s stockholders, claiming that in connection with the facts and circumstances underlying the allegations in the SEC Complaint, the Company engaged in fraudulent conduct and made false and misleading statements of material fact or omitted to state material facts necessary to make the statements made not misleading. The Sharon Claim sought both to declare the action a class action and monetary damages. The Court closed this case pending resolution of the U.S.-based class actions relating to the allegations in the SEC Complaint. The U.S. class action lawsuit was settled, and the damages granted in that settlement are for both NASDAQ and TASE class members (the “ Sharon Settlement Agreement”), Sharon filed a motion and request that the Court order the TASE to provide the information required to allow distribution of payment to TASE class members, in accordance with the Sharon Settlement Agreement.
On or about September 16, 2018, Dalia Avraham filed an Application for Approval of a Class Action in the Tel Aviv Israel District Court against the Company and Dr. Frost. This application was filed by a purported stockholder, both individually and on behalf of a putative class of the Company’s stockholders (the “Avraham Claim”). The Avraham Claim alleged a negligent and/or deliberate act related to the trade of the Company’s shares on the Tel Aviv Stock Exchange (“TASE”) which was intended to or which in fact caused damage to the Company’s investors based on the Company’s decision to delist from TASE in April 2018 and its subsequent decision to continue to be listed on TASE. The Avraham Claim sought to declare the action to be a class action and an estimated NIS 20 million (approximately USD $6.1 million) in damages. On June 24, 2021, the Court granted the parties' joint motion to withdraw and dismissed the case, subject to the payment of NIS 45,000 (approximately USD $14,200) to the plaintiff's counsel and certain other minor payments, which the Company has paid.
On October 2, 2018 Andy Yu (“Yu”), a purported stockholder, filed a shareholder derivative complaint in the United States District Court for the Southern District of Florida against the Company as a nominal defendant, certain of the Company’s current and former executive officers, certain current and former members of its Board of Directors, and Frost Gamma Investments Trust (the “Yu Lawsuit”). The Yu Lawsuit alleged violations of the federal securities laws based on alleged false and misleading statements of material fact or omissions, breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, and unjust enrichment. The Yu Lawsuit sought to direct the Company to improve its corporate governance and internal procedures, monetary damages, restitution, an award of reasonable attorneys’ fees and expert fees and other costs, and any other relief the Court might have deemed just and proper. Following the Settlement, the Yu Lawsuit was dismissed with prejudice on January 13, 2021.
On October 31, 2018, Lisette Demetriades (“Demetriades”), a purported stockholder, filed a shareholder derivative complaint in the United States District Court for the Southern District of Florida against the Company as a nominal defendant, certain of the Company’s current and former executive officers, certain current and former members of its Board of Directors, and Frost Gamma Investment Trust (the “Demetriades Lawsuit”). The Demetriades Lawsuit alleged violations of the federal securities laws based on alleged false and misleading statements of material fact or omissions, breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, and unjust enrichment. Demetriades sought to direct the Company to improve its corporate governance and internal procedures, monetary damages, restitution, an award of reasonable attorneys’ fees and expert fees and other costs, and any other relief the Court might have deemed just and proper. Following the Settlement, a Joint Stipulation for Dismissal with Prejudice was filed on January 5, 2021 and the Demetriades Lawsuit was dismissed.
On September 27, 2018, Frank Lipsius (“Lipsius”), a purported stockholder, filed a shareholder derivative complaint in the Circuit Court of the Eleventh Judicial Circuit in and for Miami-Dade County, Florida against the Company as a nominal
54
defendant, certain of the Company’s current and former executive officers, and members of its Board of Directors (the “Lipsius Lawsuit”). This lawsuit was brought by Lipsius and alleged breach of fiduciary duty against the officers and directors named therein, based on the allegations raised by the SEC in the SEC Lawsuit that the Company made misleading statements, and based on a purported failure to maintain proper internal controls. Lipsius sought to direct the Company to improve its corporate governance and internal procedures, monetary damages, restitution, an award of reasonable attorneys’ fees and expert fees and other costs, and any other relief the Court might have deemed just and proper. Following the Settlement, a Joint Stipulation for Dismissal with Prejudice was filed on December 18, 2020 and the Lipsius Lawsuit was dismissed.
On or about November 2, 2018, Louis T. Alexander (“Alexander”), a purported stockholder, filed a shareholder derivative complaint in the Circuit Court of the Eleventh Judicial Circuit of Florida serving Miami-Dade County against the Company, as a nominal defendant, Dr. Frost, certain current and former members of the Company’s Board of Directors and executive officers (the “Alexander Lawsuit”). This lawsuit alleged breach of fiduciary duty against the officers and directors named therein, based on the allegations raised by the SEC in the SEC Lawsuit that the Company made misleading statements, and based on a purported failure to maintain proper internal controls. The Alexander Lawsuit sought to direct the Company to improve its corporate governance and internal procedures, monetary damages, restitution, an award of reasonable attorneys’ fees, and experts’ fees, and other costs, and any other relief the Court might have deemed just and proper. Following the Settlement, a Joint Stipulation for Dismissal with Prejudice was filed on December 21, 2020 and the Alexander Lawsuit was dismissed.
On November 14, 2018, Sammy Lee (“Lee”), a purported stockholder, filed a shareholder derivative complaint in the United States District Court for the Southern District of Florida against the Company as a nominal defendant, Dr. Frost, Frost Gamma Investments Trust, an entity controlled by Dr. Frost, and certain of the Company’s current and former executive officers, and current and former members of its Board of Directors (the “Lee Lawsuit”). The Lee Lawsuit alleged violations of the federal securities laws based on alleged false and misleading statements of material fact or omissions, breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, and unjust enrichment. Lee sought to direct the Company to improve its corporate governance and internal procedures, monetary damages, restitution, an award of reasonable attorneys’ fees and expert fees and other costs, and any other relief as Court might have deemed just and proper. The action was dismissed sua sponte by the Judge for failure to state a claim. An amended complaint was filed on November 30, 2018. Following the Settlement, a Joint Stipulation for Dismissal with Prejudice was filed and the Lee Lawsuit was dismissed on January 11, 2021.
On December 17, 2018, Thaddeus R. Sobieski and Donnis W. King, purported stockholders of the Company, filed a shareholder derivative complaint in the United States District Court for the Southern District of Florida against the Company as a nominal defendant, Dr. Frost, Frost Gamma Investments Trust, and certain of the Company’s current and former executive officers, and current and former members of its Board of Directors (the “Sobieski King Lawsuit”). The Sobieski King Lawsuit alleged breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, violations of the federal securities laws based on alleged false and misleading statements of material fact or omissions, unjust enrichment, and waste of corporate assets. The lawsuit sought to declare that the action was a proper derivative action, a declaration that defendants breached their fiduciary duties, monetary damages, current and former directors to remit all salaries and other compensation received during the period of alleged breach of fiduciary duties, to have the Company improve its corporate governance and internal procedures, an award of pre-judgement and post- judgement interest, reasonable attorneys’ fees, experts’ fees, costs and expenses, and any other relief the Court might have deemed just and proper. Following the Settlement, the Sobieski King Lawsuit was dismissed with prejudice on January 7, 2021.
On December 31, 2018, Connie Wendt (“Wendt”), a purported stockholder, filed a shareholder derivative complaint in the United States District Court for the Southern District of Florida against the Company as a nominal defendant, certain of its current and former executive officers, certain current and former members of its Board of Directors, Frost Gamma Investments Trust, and certain other individuals and an entity named in the SEC Complaint (the “Wendt Lawsuit”). The Wendt Lawsuit alleged breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, breach of duty of loyalty, breach of fiduciary duty for insider selling and misappropriation of information, unjust enrichment, and violations of the federal securities laws, and sought a declaration that each of the Company’s named officers and directors breached their fiduciary duties to the Company, monetary damages, disgorgement of alleged profits, an award of costs and disbursements including reasonable attorneys’ fees, accountants’ and experts’ fees and costs and expenses, and any other relief the Court might have deemed just and proper. Following the Settlement, the Wendt Lawsuit was dismissed with prejudice on December 18, 2020.
On January 28, 2019, Robert Davydov (“Davydov”), a purported stockholder, filed a shareholder derivative complaint in the Circuit Court of the Eleventh Judicial Circuit of Florida serving Miami-Dade County against the Company as a nominal defendant and the certain members of its Board of Directors (the “Davydov Lawsuit”). The Davydov Lawsuit alleged breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, breach of the duties of care, loyalty and good faith, corporate waste and unjust enrichment. Davydov sought a declaration that the defendants breached their fiduciary duties,
55
an award of monetary damages, restitution, an award of reasonable attorneys’ fees and expert witness fees and other costs, and such other relief as the Court might have deemed just and proper. Following the Settlement, a Joint Stipulation for Dismissal with Prejudice was filed on December 15, 2020 and the Davydov Lawsuit was dismissed.
Between October 2018 and January 2019, five shareholder derivative complaints were filed by purported stockholders in the Delaware Court of Chancery against the Company as a nominal defendant, Dr. Frost and the Company’s Board of Directors. In the first quarter of 2019, the five lawsuits were consolidated into In re OPKO Health, Inc. Derivative Action, Case No. 2018-0740-SG (the “Consolidated Delaware Action”). On February 11, 2019, a Verified Consolidated Derivative Complaint was filed in the Consolidated Delaware Action, and Richard Tunick, Jamie Gewirtz, Emily Gewirtz Stiebel, Esther Susan Lutzker and Ivan Pawlenko were named as Co-Lead Plaintiffs in the filing. The Consolidated Derivative Complaint alleged breach of fiduciary duty based on the allegations raised by the SEC in the SEC Complaint, and breach of the duty of loyalty. The lead plaintiffs sought to declare the action a proper derivative action, monetary damages, disgorgement of all remuneration during the relevant time period, to direct the Company to improve its internal controls, equitable and injunctive relief, pre- and post-judgment interest, an award of reasonable attorneys’ fees and expert fees, and such other relief as the Court deemed just and proper. The Settlement was approved on November 2, 2020.
As previously reported, BioReference receives and is routinely required to respond to Civil Investigative Demands (“CID”) in the ordinary course of business. On November 26, 2019, BioReference received a CID from the U.S. Department of Justice (“DOJ”). The CID states that DOJ is investigating whether BioReference paid unlawful remuneration to health care practitioners in violation of the Anti-Kickback Statute or Stark law and thus submitted or caused to be submitted false claims to government health care programs in violation of the False Claims Act. The time period covered by DOJ’s requests is January 1, 2011 through November 26, 2019. BioReference has fully cooperated with the DOJ by submitting the requested information and making current employees available for interviews, and DOJ recently made a presentation to BioReference regarding its position. The parties have reached verbal agreement on the settlement amount, which is anticipated to be approximately $10 million, excluding attorney fees.
On March 1, 2019, the Company received a Civil Investigative Demand (“CID”) from the U.S. Department of Justice, Washington, DC. The CID sets forth document requests and interrogatories in connection with allegations that the Company and certain of its affiliates violated the False Claims Act and/or the Anti-Kickback Statute. On January 13, 2022, the Federal Government notified the U.S.D.C., Middle District Florida, Jacksonville Division, that it is declining to intervene in the matter but retains the right, via the Attorney General, to consent to any proposed dismals of the action by the Court. On February 9, 2022, the States of Florida, Georgia, and Commonwealth of Massachusetts notified the U.S.D.C., Middle District Florida, Jacksonville Division, that they are declining to intervene in the matter. Notwithstanding the above declinations, on February 17, 2022, the Company was served with the Relator’s Summons and Complaint (“Complaint”), which had been previously sealed. The complaint alleges violations of the False Claims Act, the California Fraud Preventions Act, the Florida False Claims Act, the Massachusetts False Claims Act, the Georgia False Medicaid Claims Act, and illegal kickbacks. The Company is reviewing and assessing the allegations made in the Complaint and, at this point, has not determined whether there is any merit to these claims nor can it determine the extent of any potential liability. While management cannot predict the outcome of these matters at this time, the ultimate outcome could be material to our business, financial condition, results of operations, and cash flows.
On April 8, 2019, MabVax Therapeutics Holdings, Inc. filed a lawsuit in the Superior Court of California, County of San Diego against a number of individuals and entities, including the Company, Dr. Frost, Steven Rubin, the Company’s Executive Vice President-Administration, and an entity affiliated with Dr. Frost, based on the allegations raised in the SEC Complaint. The lawsuit seeks an award for actual and punitive damages, pre- and post-judgment interest; that the defendants be required to make full disclosure and accounting of their interests and transactions in plaintiff’s securities; costs of the suit, and reasonable attorney’s fees; and such other legal and equitable relief as the Court may deem proper under the circumstances. On January 31, 2022, plaintiffs entered into a confidential mutual release and settlement agreement with the Company, Dr. Frost, Frost Gamma Investment Trust, and Steve Rubin (the “Settlement Agreement”). The Settlement Agreement is subject to the approval of United States Bankruptcy Court for the District of Delaware.
On April 5, 2019, former shareholders of Claros Diagnostics, Inc. filed a complaint in the Chancery Court of Delaware against the Company, alleging among other things, that the Company breached the Agreement and Plan of Merger dated October 13, 2011 by and among the Company, Claros Merger Subsidiary, LLC and Claros Diagnostics, Inc. (the “Merger Agreement”): (i) by failing to make a milestone payment of $2.375 million (payable in OPKO Common Stock) upon obtaining FDA approval of the Claros PSA test; and (ii) by repudiating its obligations to make additional future milestone payments as required under the Merger Agreement. In January 2021, the Company and the shareholder representative entered into a settlement agreement providing, among other things, that it pay the shareholders $1.1875 million, which amount is equal to half the initial milestone payable under the Merger Agreement. A Stipulation of Dismissal With Prejudice was filed with the Court on February 8, 2021.
56
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
57
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES.
Our Common Stock is traded publicly on the NASDAQ Stock Market (“NASDAQ”) and the Tel Aviv Stock Exchange under the symbol “OPK”.
As of February 25, 2022, there were approximately 363 holders of record of our Common Stock.
We have not declared or paid any cash dividends on our Common Stock. No cash dividends have been previously paid on our Common Stock and none are anticipated in fiscal 2022. We repurchased no shares of Common Stock during the fourth quarter of the year ended December 31, 2021.
Stock Performance Graph
The following graph compares the five-year cumulative total return of our Common Stock with the S&P 500 Index and the NASDAQ Biotechnology Index. The graph assumes $100 invested on December 31, 2016 in our Common Stock and in each of the foregoing indices. The stock price performance reflected in the graph below is not necessarily indicative of future price performance.
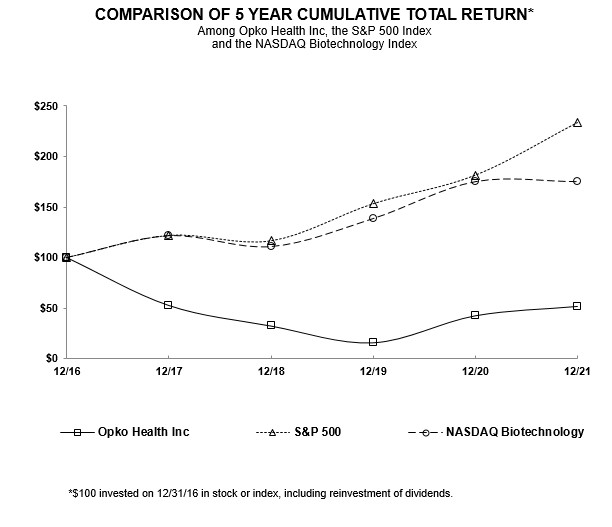
| 12/31/2016 | 12/31/2017 | 12/31/2018 | 12/31/2019 | 12/31/2020 | 12/31/2021 | ||||||||||||||||||||||||||||||
| OPKO Health, Inc. | $ | 100.00 | $ | 52.69 | $ | 32.37 | $ | 15.81 | $ | 42.47 | $ | 51.72 | |||||||||||||||||||||||
| S&P 500 | 100.00 | 121.83 | 116.49 | 153.17 | 181.35 | 233.41 | |||||||||||||||||||||||||||||
| NASDAQ Biotechnology | 100.00 | 121.63 | 110.85 | 138.69 | 175.33 | 175.37 | |||||||||||||||||||||||||||||
Recent Sales of Unregistered Securities
All recent sales of unregistered securities were previously reported in a Current Report on Form 8-K or Quarterly Report on Form 10-Q.
ITEM 6. Selected Financial Data.
Not applicable.
59
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 (“PSLRA”), Section 27A of the Securities Act of 1933, as amended, (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended, (the “Exchange Act”), about our expectations, beliefs, or intentions regarding our product development efforts, business, financial condition, results of operations, strategies and prospects. You can identify forward-looking statements by the fact that these statements do not relate to historical or current matters. Rather, forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements. Many factors could cause our actual activities or results to differ materially from the activities and results anticipated in forward-looking statements. These factors include those contained in “Item 1A — Risk Factors” of this Annual Report on Form 10-K. We do not undertake any obligation to update forward-looking statements except as required by applicable law. We intend that all forward-looking statements be subject to the safe harbor provisions of PSLRA. These forward-looking statements reflect our views only as of the date they are made.
OVERVIEW
We are a diversified healthcare company that seeks to establish industry-leading positions in large and rapidly growing medical markets. Our diagnostics business includes BioReference Laboratories, Inc. (“BioReference”), one of the nation’s largest full service laboratories with an almost 250-person sales and marketing team to drive growth and leverage new products. Our pharmaceutical business features Rayaldee, a U.S. Food and Drug Administration (“FDA”) approved treatment for secondary hyperparathyroidism (“SHPT”) in adults with stage 3 or 4 chronic kidney disease (“CKD”) and vitamin D insufficiency and a pipeline of products in various stages of development. Our leading product in development is Somatrogon (hGH-CTP), a once-weekly human growth hormone for which we have partnered with Pfizer, Inc. (“Pfizer”) and successfully completed a phase 3 study in August 2019. Regulatory applications for Somatrogon have been submitted to several countries around the world for review. In February 2022, the European Commission granted marketing authorization in the European Union for Somatrogon under the brand name NGENLA® to treat children and adolescents from as young as 3 years of age with growth disturbance due to insufficient secretion of growth hormone. In January 2022, the Ministry of Health, Labour and Welfare in Japan approved NGENLA® (Somatrogon) for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. In October 2021, Health Canada approved NGENLA® for the long-term treatment of pediatric patients who have growth hormone deficiency, and Australia’s Therapeutic Goods Administration approved NGENLA® for the long-term treatment of pediatric patients with growth disturbance due to insufficient secretion of growth hormone. We also submitted the initial Biologics License Application (“BLA”) with the FDA for approval of Somatrogon (hGH-CTP) in the United States and Pfizer received a Complete Response Letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States. We are incorporated in Delaware, and our principal executive offices are located in leased offices in Miami, Florida.
Through BioReference, we provide laboratory testing services, primarily to customers in the larger metropolitan areas in New York, New Jersey, Florida, Texas, Maryland, California, Pennsylvania, Delaware, Washington, DC, Illinois and Massachusetts, as well as to customers in a number of other states. We offer a comprehensive test menu of clinical diagnostics for blood, urine and tissue analysis. This includes hematology, clinical chemistry, immunoassay, infectious diseases, serology, hormones, and toxicology assays, as well as Pap smear, anatomic pathology (biopsies) and other types of tissue analysis. We market our laboratory testing services directly to physicians, geneticists, hospitals, clinics, correctional and other health facilities.
We operate established pharmaceutical platforms in Ireland, Chile, Spain, and Mexico, which are generating revenue and from which we expect to generate positive cash flow and facilitate future market entry for our products currently in development. In addition, we have a development and commercial supply pharmaceutical company and a global supply chain operation and holding company in Ireland. We own a specialty active pharmaceutical ingredients manufacturer in Israel, which we expect will facilitate the development of our pipeline of molecules and compounds for our proprietary molecular diagnostic and therapeutic products.
RECENT DEVELOPMENTS
In early 2022, each of the European Commission and the Ministry of Health, Labour and Welfare in Japan approved the next-generation long-acting recombinant human growth hormone NGENLA (Somatrogon), a once-weekly injection to treat
60
pediatric growth hormone deficiency in Europe and Japan, respectively. Further, Canada and Australia approved NGENLA in October and November of 2021, respectively.
In January 2022, the FDA issued a Complete Response Letter for the BLA for Somatrogon. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States.
In January 2022, Sema4 Holdings Corp. (“Sema4”) and OPKO announced they had signed a definitive agreement (the “GeneDx Merger Agreement”) pursuant to which Sema4 has agreed to acquire OPKO’s wholly owned subsidiary, GeneDx, Inc. (“GeneDx”), a leader in genomic testing and analysis, subject to the satisfaction of customary closing conditions (the “GeneDx Transaction”). The GeneDx Transaction is expected to close in the second quarter of 2022.
Under the terms of the GeneDx Merger Agreement, Sema4 has agreed to acquire GeneDx for an upfront payment of $150 million in cash together with 80.0 million shares of Sema4’s Class A common stock, par value $0.0001 per share (“Sema4 Common Stock”), subject to a customary purchase price adjustment mechanism providing for a normalized level of working capital and that GeneDx be free of debt at closing of the GeneDx Transaction. Additionally, Sema4 agreed to pay OPKO up to an additional $150.0 million, which may be paid in Sema4 Common Stock, cash or a combination thereof in Sema4’s discretion, subject to GeneDx achieving certain revenue targets for the fiscal years ending December 31, 2022 and 2023. Based on the closing stock price of Sema4 Common Stock as of January 14, 2022, the total upfront consideration is approximately $473 million, and the total aggregate consideration including potential milestones is approximately $623 million.
As of December 31, 2021, GeneDx met the held-for-sale accounting criteria and the related assets and liabilities are classified as held for sale in the consolidated balance sheet. Depending upon the value Sema4 shares upon closing of the transaction, an impairment charge may be incurred. GeneDx was included in our diagnostics segment as of December 31, 2021.
In December 2021, we announced preliminary topline results from our Phase 2 trial with Rayaldee to treat mild-to-moderate COVID-19 which indicate that vitamin D repletion therapy can accelerate recovery from COVID-19.
In December 2021, we announced that the FDA approved our 4Kscore test for use in men age 45 and older who have not had a prior prostate biopsy or are biopsy negative and have an age-specific abnormal total PSA and/or abnormal digital rectal exam.
RESULTS OF OPERATIONS
Impact of COVID-19
As the disease caused by SARS-CoV-2, a novel strain of coronavirus, COVID-19 continues to spread and severely impact the U.S. economy and economies of other countries around the world, we continue to be a part of the coordinated public and private sector response to this unprecedented challenge. There continues to be a high level of uncertainty relating to how the pandemic will evolve, how governments and consumers will react, progress on the distribution of vaccines and whether the pandemic will have a longer-term effect on the healthcare industry and patient habits. In response to the COVID-19 pandemic, BioReference is providing COVID-19 solutions, including diagnostic molecular testing and serology antibody testing, to meet the testing needs of its customers, including physicians, health systems, long-term care facilities, governments, schools, employers, professional sports teams and entertainment venues, as well as the general public through relationships with retail pharmacy chains.
Revenue from services for the year ended December 31, 2021 increased by $344.9 million as compared to 2020 due to COVID-19 testing volumes. We are unable to predict how long the demand will continue for our COVID-19 related testing, or whether pricing and reimbursement policies for testing will be sustained. In addition, in the second half of 2021, overall demand for COVID-19 testing declined. Additionally, beginning in March 2020, BioReference experienced a decline in testing volumes due to the COVID-19 pandemic; however as stay at home orders and other restrictions have been lifted, we have seen our routine clinical testing volumes trending towards normalization with prior periods. Should stay at home orders or other restrictions be reenacted, we could see our routine testing levels decline. Excluding COVID-19 test volumes, for the year ended December 31, 2021, genomic and routine clinical test volume increased 26.4% and 6.9%, respective, as compared to volumes for the year ended December 31, 2020. Additionally, sales of Rayaldee have not increased in accordance with its expected growth trajectory as a result of challenges in onboarding new patients due to the COVID-19 pandemic. Federal, state and local governmental policies and initiatives designed to reduce the transmission of COVID-19 have resulted in, among other things, a significant reduction in physician office visits, the cancellation of elective medical procedures, customers closing or severely
61
curtailing their operations (voluntarily or in response to government orders), and the adoption of work-from-home or shelter-in-place policies.
In March 2020, in response to the COVID-19 pandemic, the Coronavirus Aid, Relief, and Economic Security (CARES) Act was signed into law. The CARES Act provides numerous tax provisions and other stimulus measures, including temporary changes regarding the prior and future utilization of net operating losses, temporary changes to the prior and future limitations on interest deductions, temporary suspension of certain payment requirements for the employer portion of Social Security taxes, technical corrections from prior tax legislation for tax depreciation of certain qualified improvement property, and the creation of certain payroll tax credits associated with the retention of employees.
We have received, or expect to receive a number of benefits under the CARES Act including, but not limited to:
•During the year ended December 31, 2020, we received approximately $14 million under The Centers for Medicare & Medicaid Services (CMS) Accelerated and Advance Payment Program, which provides accelerated payments to Medicare providers/suppliers working to provide treatment to patients and combat the COVID-19 pandemic, and the amounts advanced are loans which will be offset against future claims and were repaid in 2021. These loans were initially recorded as contract liabilities included in Accrued expenses and were reduced as the amounts are recouped by CMS;
•We are eligible to defer depositing the employer’s share of Social Security taxes for payments due from March 27, 2020 through December 31, 2020, interest-free and penalty-free;
•We received approximately $16.2 million during 2020 from the funds that were distributed to healthcare providers for related expenses or lost revenues that are attributable to the COVID-19 pandemic. We recognized the $16.2 million grant in other revenues for the year ended December 31, 2020;
•U.S. Department of Health and Human Services (HHS), will provide claims reimbursement to healthcare providers generally at Medicare rates for testing uninsured patients; and
•Clinical laboratories are provided a one-year reprieve from the reporting requirements under PAMA as well as a one-year delay of reimbursement rate reductions for clinical laboratory services provided under Medicare that were scheduled to take place in 2021.
Since the pandemic began in the U.S., we have invested in testing capabilities and infrastructure to meet demand for our molecular and antibody testing for COVID-19. In 2021, we kicked off company-wide lab operations specimen acquisition, logistics, procurement, customer service, cost reduction initiatives to right size our cost structure to match the declining COVID testing volumes and to drive efficiency gains in our core clinical lines of business.
Three vaccines for COVID-19 have received approval or emergency authorization and have had increasingly widespread acceptance. However, we believe that, based on our experience with the pandemic, the high medical need for efficient and widespread testing for COVID-19 will extend beyond the current phase of the pandemic. Our belief is supported by the unprecedented healthcare and economic impact of the pandemic thus far, the uneven and incomplete rollout of vaccines and the fact that significant portions of the U.S. population may never be vaccinated, and the continued likelihood of surges of COVID-19 including from new strains of SARS-CoV-2 with uncertain susceptibility to the current vaccines. We believe that these factors have greatly magnified the need for more effective therapeutics, and the need for efficient and widespread testing.
For The Years Ended December 31, 2021 and December 31, 2020
Our consolidated income (loss) from operations for the years ended December 31, 2021 and 2020 is as follows:
62
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2021 | 2020 | Change | % Change | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Revenue from services | $ | 1,607,106 | $ | 1,262,242 | $ | 344,864 | 27 | % | ||||||||||||
| Revenue from products | 141,770 | 119,952 | 21,818 | 18 | % | |||||||||||||||
| Revenue from transfer of intellectual property and other | 25,842 | 53,219 | (27,377) | (51) | % | |||||||||||||||
| Total revenues | 1,774,718 | 1,435,413 | 339,305 | 24 | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 1,193,194 | 894,408 | 298,786 | 33 | % | |||||||||||||||
| Selling, general and administrative | 468,857 | 355,573 | 113,284 | 32 | % | |||||||||||||||
| Research and development | 76,850 | 75,316 | 1,534 | 2 | % | |||||||||||||||
| Contingent consideration | (1,703) | (3,989) | 2,286 | (57) | % | |||||||||||||||
| Amortization of intangible assets | 50,278 | 56,391 | (6,113) | (11) | % | |||||||||||||||
| Gain on sale of assets | (31,508) | — | (31,508) | (100) | % | |||||||||||||||
| Total costs and expenses | 1,755,968 | 1,377,699 | 378,269 | 27 | % | |||||||||||||||
| Income (loss) from operations | 18,750 | 57,714 | (38,964) | (68) | % | |||||||||||||||
Diagnostics
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2021 | 2020 | Change | % Change | ||||||||||||||||
| Revenues | ||||||||||||||||||||
| Revenue from services | $ | 1,607,106 | $ | 1,262,242 | 344,864 | 27 | % | |||||||||||||
| Revenue from transfer of intellectual property and other | — | 16,240 | (16,240) | (100) | % | |||||||||||||||
| Total revenues | 1,607,106 | 1,278,482 | 328,624 | 26 | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 1,102,175 | 823,927 | 278,248 | 34 | % | |||||||||||||||
| Selling, general and administrative | 357,633 | 266,488 | 91,145 | 34 | % | |||||||||||||||
| Research and development | 18,652 | 15,003 | 3,649 | 24 | % | |||||||||||||||
| Contingent consideration | — | (2,066) | 2,066 | (100) | % | |||||||||||||||
| Amortization of intangible assets | 30,579 | 36,208 | (5,629) | (16) | % | |||||||||||||||
| Total costs and expenses | 1,509,039 | 1,139,560 | 369,479 | 32 | % | |||||||||||||||
| Income from operations | 98,067 | 138,922 | (40,855) | (29) | % | |||||||||||||||
Revenue. Revenue from services for the year ended December 31, 2021 increased by approximately $344.9 million compared to the year ended December 31, 2020, due to an improvement in clinical test reimbursement, and an increase in clinical test volume and genomic test volume of $38.1 million, $33.5 million and $25.1 million, respectively. This was partially offset by the negative impact of a reduction in genomic test reimbursement of $5.6 million.
BioReference also recognized an increase in revenue for the year ended December 31, 2021 compared to the year ended December 31, 2020 due to an increase in COVID-19 testing volume and improvement in COVID-19 test reimbursement of $97.0 million and $141.4 million, respectively. BioReference performed 11.9 million diagnostic molecular tests for COVID-19 and 0.7 million serology antibody tests during the year ended December 31, 2021, which represented 58.5% of total test volume for that period. In comparison, during the year ended December 31, 2020, BioReference performed 10.2 million molecular tests for COVID-19 and 0.8 million serology antibody tests.
Estimated collection amounts are subject to the complexities and ambiguities of billing, reimbursement regulations and claims processing, as well as considerations unique to Medicare and Medicaid programs, and require us to consider the potential for retroactive adjustments when estimating variable consideration in the recognition of revenue in the period the related services are rendered. Revenue from services for the year ended December 31, 2020 included $12.1 million related to
63
the successful appeal of previously denied claims for the 4Kscore test. In addition, the years ended December 31, 2021 and 2020 included positive revenue adjustments due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $40.4 million and $0.3 million, respectively, were recognized.
The composition of revenue from services by payor for the years ended December 31, 2021 and 2020 was as follows:
| For the years ended December 31, | |||||||||||
| (In thousands) | 2021 | 2020 | |||||||||
| Healthcare insurers | $ | 520,244 | $ | 483,643 | |||||||
| Government payers | 222,242 | 90,288 | |||||||||
| Client payers | 843,405 | 637,645 | |||||||||
| Patients | 21,215 | 50,666 | |||||||||
| Total | $ | 1,607,106 | $ | 1,262,242 | |||||||
Client payors include cities, states and companies for which BioReference provides COVID-19 testing services.
Revenue from transfer of intellectual property and other for the year ended December 31, 2020 are the result of grants received under the CARES Act totaling $16.2 million.
Cost of revenue. Cost of revenue for the year ended December 31, 2021 increased $278.2 million compared to the year ended December 31, 2020. Cost of revenue increased primarily due to labor and material costs for COVID-19 testing and the significant volume of tests performed during the year ended December 31, 2021. Cost of revenue for the year ended December 31, 2021 also increased due to changes in the product mix of items sold during the period, which was partially offset by a $5.5 million sales and use tax credit received during the year ended December 31, 2021.
Selling, general and administrative expenses. Selling, general and administrative expenses for the years ended December 31, 2021 and 2020 were $357.6 million and $266.5 million, respectively. Selling, general and administrative expenses in our diagnostics segment increased primarily due to higher variable billing and compensation costs which resulted from an increase in volume and collections during the year ended December 31, 2021, and in marketing costs and other administrative costs directly associated with COVID-19 testing volumes. Selling, general and administrative expenses for the year ended December 31, 2021 also include $6.2 million of expense incurred in connection with certain legal matters and $40.0 million in administrative, IT, and marketing costs associated with our investment in the launch of Scarlet Health.
Research and development expenses. The following table summarizes the components of our research and development expenses:
| Research and Development Expenses | For the years ended December 31, | ||||||||||
| 2021 | 2020 | ||||||||||
| External expenses: | |||||||||||
| PMA studies | $ | — | $ | 218 | |||||||
| Research and development employee-related expenses | 13,266 | 9,317 | |||||||||
| Other internal research and development expenses | 5,386 | 5,468 | |||||||||
| Total research and development expenses | $ | 18,652 | $ | 15,003 | |||||||
The increase in research and development expenses for the year ended December 31, 2021 resulted primarily from increased research and development expenses related to the development of clinical and genomics testing services.
Contingent consideration. Contingent consideration for the years ended December 31, 2021 and 2020 was $0 thousand and $2.1 million reversal of expense, respectively. Contingent consideration for the year ended December 31, 2020 was attributable to changes in assumptions regarding the timing of achievement of future milestones for OPKO Diagnostics, and potential amounts payable to former stockholders of OPKO Diagnostics in connection therewith, pursuant to our acquisition agreement in October 2011.
Amortization of intangible assets. Amortization of intangible assets was $30.6 million and $36.2 million, for the years ended December 31, 2021 and 2020, respectively. Amortization expense reflects the amortization of acquired intangible assets with defined useful lives. Amortization expense declined during the year ended December 31, 2021 due to acquired intangible assets becoming fully amortized.
64
Pharmaceuticals
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2021 | 2020 | Change | % Change | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Revenue from products | $ | 141,770 | $ | 119,952 | $ | 21,818 | 18 | % | ||||||||||||
| Revenue from transfer of intellectual property and other | 25,842 | 36,979 | (11,137) | (30) | % | |||||||||||||||
| Total revenues | 167,612 | 156,931 | 10,681 | 7 | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 91,059 | 70,565 | 20,494 | 29 | % | |||||||||||||||
| Selling, general and administrative | 49,375 | 50,476 | (1,101) | (2) | % | |||||||||||||||
| Research and development | 59,741 | 61,149 | (1,408) | (2) | % | |||||||||||||||
| Contingent consideration | (1,703) | (1,923) | 220 | (11) | % | |||||||||||||||
| Amortization of intangible assets | 19,699 | 20,183 | (484) | (2) | % | |||||||||||||||
| Gain on sale of asset | (31,508) | — | (31,508) | (100) | % | |||||||||||||||
| Total costs and expenses | 186,663 | 200,450 | (13,787) | (7) | % | |||||||||||||||
| Loss from operations | (19,051) | (43,519) | 24,468 | (56) | % | |||||||||||||||
Revenue. The increase in revenue from products for the year ended December 31, 2021 compared to the year ended December 31, 2020 was primarily attributable to an increase in sales at most of our international operating companies. Sales of Rayaldee were $27.0 million for the year ended December 31, 2021, as compared to $36.8 million for 2020. Sales of Rayaldee have been negatively impacted as a result of challenges in onboarding new patients due to the COVID-19 pandemic. Revenue from transfer of intellectual property for the years ended December 31, 2021 and 2020 principally reflected $10.8 million and $28.7 million, respectively, of revenue related to the Pfizer Transaction. Revenue from transfer of intellectual property and other for the year ended December 31, 2021 also included a $5.0 million non-refundable upfront payment we received under the Nicoya Agreement (as defined below) and receipt of a $4.9 million payment under the CAMP4 Agreement. Revenue from transfer of intellectual property for the year ended December 31, 2020 also included a $3.0 million milestone payment triggered by the first marketing approval of Rayaldee in Europe.
Cost of revenue. Cost of revenue for the year ended December 31, 2021 increased $20.5 million compared to the year ended December 31, 2020. Cost of product revenue increased primarily due to an increase in inventory and material costs at most of our international operating companies, which was due to the increase in sales at our international operating companies and to a $3.8 million inventory reserve recognized for Rayaldee inventory for the year ended December 31, 2021. This was partially offset by a decrease in sales of Rayaldee for the year ended December 31, 2021 compared to the year ended December 31, 2020.
Selling, general and administrative expenses. Selling, general and administrative expenses for the years ended December 31, 2021 and 2020 were $49.4 million and $50.5 million, respectively. Selling, general and administrative expenses for the year ended December 31, 2021 was consistent with selling, general and administrative expenses for the year ended December 31, 2020. Selling, general and administrative expenses for the pharmaceutical segment for the years ended December 31, 2021 and 2020 included equity-based compensation expense of $1.3 million and $1.0 million, respectively.
Research and development expenses. Research and development expenses for the years ended December 31, 2021 and 2020 were $59.7 million and $61.1 million, respectively. Research and development expenses include external and internal expenses, partially offset by third-party grants and funding arising from collaboration agreements. External expenses include clinical and non-clinical activities performed by contract research organizations, lab services, purchases of drug and diagnostic product materials and manufacturing development costs. We track external research and development expenses by individual program for phase 3 clinical trials for drug approval and premarket approval for diagnostics tests, if any. Internal expenses include employee-related expenses such as salaries, benefits and equity-based compensation expense. Other internal research and development expenses are incurred to support overall research and development activities and include expenses related to general overhead and facilities.
The following table summarizes the components of our research and development expenses:
65
| Research and Development Expenses | For the years ended December 31, | ||||||||||
| 2021 | 2020 | ||||||||||
| External expenses: | |||||||||||
| Manufacturing expense for biological products | $ | 9,718 | $ | 5,326 | |||||||
| Phase III studies | 9,877 | 10,513 | |||||||||
| Post-marketing studies | 70 | 1,270 | |||||||||
| Earlier-stage programs | 17,268 | 14,811 | |||||||||
| Research and development employee-related expenses | 19,497 | 21,931 | |||||||||
| Other internal research and development expenses | 3,614 | 9,440 | |||||||||
| Third-party grants and funding from collaboration agreements | (303) | (2,142) | |||||||||
| Total research and development expenses | $ | 59,741 | $ | 61,149 | |||||||
Research and development expenses for the year ended December 31, 2021 was consistent with research and development expenses for the year ended December 31, 2020. Research and development expenses for the year ended December 31, 2021 were primarily due to expenses related to Somatrogon, a once-weekly human growth hormone injection for which we have partnered with Pfizer. Ongoing expenses for the Somatrogon program support open label extension studies that will continue until the market launch of Somatrogon in certain countries, as well as the preparation of applications for marketing approvals. Research and development expenses for the pharmaceutical segment for the years ended December 31, 2021 and 2020 included equity-based compensation expense of $1.3 million and $1.6 million, respectively.
Contingent consideration. Contingent consideration for the years ended December 31, 2021 and 2020 was $1.7 million and $1.9 million reversal of expense, respectively. Contingent consideration for the year ended December 31, 2021 was primarily attributable to changes in assumptions regarding the timing of achievement of future milestones for OPKO Renal and OPKO CURNA, and potential amounts payable to former stockholders of OPKO Renal and OPKO CURNA in
connection therewith, pursuant to our acquisition agreements in March 2013 and January 2011, respectively. Contingent consideration for the year ended December 31, 2020 was primarily attributable to changes in assumptions regarding the timing of achievement of future milestones for OPKO Renal, and potential amounts payable to former stockholders of OPKO Renal.
Amortization of intangible assets. Amortization of intangible assets was $19.7 million and $20.2 million, respectively, for the years ended December 31, 2021 and 2020. Amortization expense reflects the amortization of acquired intangible assets with defined useful lives. Our indefinite lived in-process research and development (“IPR&D”) assets will not be amortized until the underlying development programs are completed. Upon obtaining regulatory approval by the FDA, the IPR&D assets will be accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life.
Gain on sale of assets. Gain on sale of assets for the year ended December 31, 2021 was $31.5 million, which resulted from an agreement between EirGen, our wholly owned subsidiary, and Horizon Therapeutics plc, to sell one of EirGen’s facilities in Waterford, Ireland for $65 million in cash less certain assumed and accrued liabilities relating to transferred employees. The facility housed EirGen’s sterile-fill-finish business and was no longer a core component of our ongoing operations and business strategy.
66
Corporate
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2021 | 2020 | Change | % Change | ||||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | $ | (40) | $ | (84) | $ | 44 | (52) | % | ||||||||||||
| Selling, general and administrative | 61,849 | 38,609 | 23,240 | 60 | % | |||||||||||||||
| Research and development | (1,543) | (836) | (707) | 85 | % | |||||||||||||||
| Total costs and expenses | 60,266 | 37,689 | 22,577 | 60 | % | |||||||||||||||
| Loss from operations | $ | (60,266) | $ | (37,689) | $ | (22,577) | 60 | % | ||||||||||||
Operating loss for our unallocated corporate operations for the years ended December 31, 2021 and 2020 was $60.3 million and $37.7 million, respectively, and principally reflects general and administrative expenses incurred in connection with our corporate operations. The increase in operating loss for the year ended December 31, 2021 was primarily attributable to an increase in legal fees incurred for the year ended December 31, 2021, compared to the year ended December 31, 2020.
Other
Interest income. Interest income for the years ended December 31, 2021 and 2020 was not significant as our cash investment strategy emphasizes the security of the principal invested and fulfillment of liquidity needs.
Interest expense. Interest expense for the years ended December 31, 2021 and 2020 was $18.9 million and $21.9 million, respectively. Interest expense was principally related to interest incurred on our Senior Convertible Notes due 2025 (the “2025 Notes”), our 5% Convertible Promissory Notes (the “2023 Convertible Notes”), our 3.0% Senior Notes due 2033 (the “2033 Senior Notes”), and BioReference’s outstanding debt under the A&R Credit Agreement.
Fair value changes of derivative instruments, net. Fair value changes of derivative instruments, net for the years ended December 31, 2021 and 2020, was $846 thousand and $50 thousand of income, respectively. Derivative income for the year ended December 31, 2021, was principally related to the change in fair value on foreign currency forward exchange contracts at OPKO Chile.
Other income (expense), net. Other income (expense), net for the years ended December 31, 2021 and 2020, was $14.8 million of expense and $12.7 million of income, respectively. Other expense for the years ended December 31, 2021 primarily consisted of a $11.1 million non-cash loss related to the exchange of $55.4 million of the outstanding 2025 Notes for 19,051,270 shares of our Common Stock and net unrealized losses recognized during the period on our investments in our equity securities. Other income for the year ended December 31, 2020 primarily consisted of realized and unrealized gains recognized during the period on our investment in VBI Vaccines Inc. (“VBI”), offset by net unrealized losses recognized during the period on our investment in Eloxx Pharmaceuticals, Inc. (“Eloxx”).
Income tax provision. Our income tax provision for the years ended December 31, 2021 and 2020 was $15.5 million and $17.6 million, respectively, and reflects results using our expected effective tax rate. For the year ended December 31, 2021, the tax rate differed from the U.S. federal statutory rate of 21% primarily due to the relative mix in earnings and losses in the U.S. versus foreign tax jurisdictions, the impact of certain discrete tax events and operating results in tax jurisdictions that do not result in a tax benefit.
Loss from investments in investees. We have made investments in certain early stage companies that we perceive to have valuable proprietary technology and significant potential to create value for us as a shareholder or member. We account for these investments under the equity method of accounting, resulting in the recording of our proportionate share of their losses until our share of their loss exceeds our investment. Until the investees’ technologies are commercialized, if ever, we anticipate they will report net losses. Loss from investments in investees was $0.6 million and $0.5 million for the years ended December 31, 2021 and 2020, respectively.
67
For The Years Ended December 31, 2020 and December 31, 2019
Our consolidated income (loss) from operations for the years ended December 31, 2020 and 2019 is as follows:
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2020 | 2019 | Change | % Change | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Revenue from services | $ | 1,262,242 | $ | 716,434 | $ | 545,808 | 76 | % | ||||||||||||
| Revenue from products | 119,952 | 112,184 | 7,768 | 7 | % | |||||||||||||||
| Revenue from transfer of intellectual property and other | 53,219 | 73,317 | (20,098) | (27) | % | |||||||||||||||
| Total revenues | 1,435,413 | 901,935 | 533,478 | 59 | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 894,408 | 572,484 | 321,924 | 56 | % | |||||||||||||||
| Selling, general and administrative | 355,573 | 343,305 | 12,268 | 4 | % | |||||||||||||||
| Research and development | 75,316 | 117,870 | (42,554) | (36) | % | |||||||||||||||
| Contingent consideration | (3,989) | (14,854) | 10,865 | (73) | % | |||||||||||||||
| Amortization of intangible assets | 56,391 | 64,783 | (8,392) | (13) | % | |||||||||||||||
| Asset impairment charges | — | 92,399 | (92,399) | (100) | % | |||||||||||||||
| Total costs and expenses | 1,377,699 | 1,175,987 | 201,712 | 17 | % | |||||||||||||||
| Income (loss) from operations | 57,714 | (274,052) | 331,766 | (121) | % | |||||||||||||||
We manage our operations in two reportable segments, pharmaceuticals and diagnostics. The pharmaceuticals segment consists of our pharmaceutical operations in Latin America, Ireland, Israel and Spain, Rayaldee product sales and our pharmaceutical research and development. The diagnostics segment primarily consists of our clinical and genetic laboratory operations through BioReference and GeneDx as well as our point-of-care operations. There are no significant inter-segment sales. We evaluate the performance of each segment based on operating profit or loss. The following presents the financial measures that management considers to be the most significant indicators of the Company's performance.
Diagnostics
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2020 | 2019 | Change | % Change | ||||||||||||||||
| Revenues | ||||||||||||||||||||
| Revenue from services | $ | 1,262,242 | $ | 716,434 | 545,808 | 76 | % | |||||||||||||
| Revenue from transfer of intellectual property and other | 16,240 | — | 16,240 | 100 | % | |||||||||||||||
| Total revenues | 1,278,482 | 716,434 | 562,048 | 78 | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 823,927 | 510,857 | 313,070 | 61 | % | |||||||||||||||
| Selling, general and administrative | 266,488 | 242,020 | 24,468 | 10 | % | |||||||||||||||
| Research and development | 15,003 | 14,219 | 784 | 6 | % | |||||||||||||||
| Contingent consideration | (2,066) | (8,401) | 6,335 | (75) | % | |||||||||||||||
| Amortization of intangible assets | 36,208 | 42,401 | (6,193) | (15) | % | |||||||||||||||
| Asset impairment charges | — | 38,697 | (38,697) | (100) | % | |||||||||||||||
| Total costs and expenses | 1,139,560 | 839,793 | 299,767 | 36 | % | |||||||||||||||
| Income (loss) from operations | 138,922 | (123,359) | 262,281 | (213) | % | |||||||||||||||
Revenue. Revenue from services for the year ended December 31, 2020 increased by approximately $545.8 million compared to the year ended December 31, 2019, due to COVID-19 testing volumes. BioReference performed 0.8 million serology antibody tests and 10.1 million diagnostic molecular tests for COVID-19 during the year ended December 31, 2020, which represented 57% of total testing volume. Revenue attributable to tests for COVID-19 was partially offset by the negative impacts of:
68
•A reduction in clinical test volumes and genomic test volumes at BioReference resulted in decreased revenues of $99.5 million and $15.6 million, respectively, as compared to the year ended December 31, 2019. The decline in routine clinical and genomic testing volume reflects negative impacts from the COVID-19 pandemic, principally from referring physician office closures and stay-at-home guidance throughout states in which we predominately operate.
•A reduction in clinical test and genomic test reimbursement at BioReference of $10.2 million and $27.6 million, respectively, as compared to the year ended December 31, 2019. The lower reimbursement within our clinical business was primarily the result of the negative impact of the PAMA price reduction that went into effect January 1, 2020 combined with an overall shift in our test mix that was partially offset by increased reimbursement of our 4KScore test. The lower reimbursement within our genomic business resulted from an increase in denial rates and changes to payor policy and procedural requirements.
Estimated collection amounts are subject to the complexities and ambiguities of billing, reimbursement regulations and claims processing, as well as considerations unique to Medicare and Medicaid programs, and require us to consider the potential for retroactive adjustments when estimating variable consideration in the recognition of revenue in the period the related services are rendered. Revenue from services for the year ended December 31, 2020 included $12.1 million related to the successful appeal of previously denied claims for the 4Kscore test. In addition, the year ended December 31, 2020 included positive revenue adjustments recognized due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $0.3 million, and for the year ended December 31, 2019, revenue reductions of $24.8 million were recognized due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods.
The composition of Revenue from services by payor for the years ended December 31, 2020 and 2019 was as follows:
| For the years ended December 31, | |||||||||||
| (In thousands) | 2020 | 2019 | |||||||||
| Healthcare insurers | $ | 483,643 | $ | 421,386 | |||||||
| Government payers | 90,288 | 115,711 | |||||||||
| Client payers | 637,645 | 158,527 | |||||||||
| Patients | 50,666 | 20,810 | |||||||||
| Total | $ | 1,262,242 | $ | 716,434 | |||||||
Client payers include cities, states and companies for which BioReference provides COVID-19 testing services.
Revenue from transfer of intellectual property and other for the year ended December 31, 2020 are the result of grants received under the CARES Act totaling $16.2 million.
Cost of revenue. Cost of revenue for the year ended December 31, 2020 increased $313.1 million compared to the year ended December 31, 2019. Cost of revenue increased primarily due to labor and material costs for COVID-19 testing and the significant volume of tests performed during the year ended December 31, 2020, partially offset by a decline in non-COVID testing volumes and to cost reduction initiatives leading to a 12.4% improvement in cost per patient encounter, inclusive of all volumes.
Selling, general and administrative expenses. Selling, general and administrative expenses for the years ended December 31, 2020 and 2019 were $266.5 million and $242.0 million, respectively. Selling, general and administrative expenses in our diagnostics segment increased primarily due to higher variable billing and compensation costs of $23.2 million from an increase in volume and collections during the year ended December 31, 2020 and $3.0 million in marketing costs and other administrative and marketing costs directly associated the COVID-19 PCR testing volumes. In comparison, the December 31, 2019 period included $12.6 million of expense related to the Department of Justice settlement. As a percentage of net revenue SG&A for the diagnostic segment decreased to 21% from 34%, for the years ended December 31, 2020 and 2019, respectively as a result of per requisition efficiencies and expense management during this recent period of rapid volume growth. Selling, general and administrative expenses for the diagnostics segment for the years ended December 31, 2020 and 2019 included equity-based compensation expense of $2.1 million and $2.2 million, respectively.
69
Research and development expenses. The following table summarizes the components of our research and development expenses:
| Research and Development Expenses | For the years ended December 31, | ||||||||||
| 2020 | 2019 | ||||||||||
| External expenses: | |||||||||||
| PMA studies | $ | 218 | $ | 774 | |||||||
| Research and development employee-related expenses | 9,317 | 7,320 | |||||||||
| Other internal research and development expenses | 5,468 | 6,125 | |||||||||
| Total research and development expenses | $ | 15,003 | $ | 14,219 | |||||||
Research and development for the diagnostic segment relates to the development of testing services for our clinical and genomics testing at BioReference and the development of the Claros Analyzer, a diagnostic instrument system to provide rapid, high performance blood test results in the point-of-care setting. The increase in research and development expenses for the year ended December 31, 2020 resulted primarily from an increased research and development expenses related to the development of clinical and genomics testing services.
Contingent consideration. Contingent consideration for the years ended December 31, 2020 and 2019 was $(2.1) million of expense and $8.4 million reversal of expense, respectively. Contingent consideration for the years ended December 31, 2020 and 2019 was attributable to changes in assumptions regarding the timing of achievement of future milestones for OPKO Diagnostics in both periods, and potential amounts payable to former stockholders of OPKO Diagnostics in connection therewith, pursuant to our acquisition agreement in October 2011.
Amortization of intangible assets. Amortization of intangible assets was $36.2 million and $42.4 million, respectively, for the years ended December 31, 2020 and 2019. Amortization expense reflects the amortization of acquired intangible assets with defined useful lives.
Asset impairment charges. Asset impairment charges were $38.7 million for the year ended December 31, 2019. Asset impairment charges for the year ended December 31, 2019 is primarily related to a goodwill impairment charge of $18.0 million to write the carrying amount of the OPKO Diagnostics reporting unit down to its estimated fair value, and an impairment charge of $20.7 million to write our intangible asset for the Claros Analyzer down to its estimated fair value. The asset impairment charges for the year ended December 31, 2019, resulted from liquidity constraints, longer than expected development timelines and changes in the competitive landscape, which resulted in changes to our estimates and assumptions of the expected future cash flows associated with the Claros Analyzer.
We believe that our estimates and assumptions in testing goodwill and other intangible assets are consistent with assumptions that marketplace participants would use in their estimates. However, if actual results are not consistent with our estimates and assumptions, including as a result of the COVID-19 global pandemic, we may be exposed to an impairment charge that could be material.
70
Pharmaceuticals
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2020 | 2019 | Change | % Change | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Revenue from products | $ | 119,952 | $ | 112,184 | $ | 7,768 | 7 | % | ||||||||||||
| Revenue from transfer of intellectual property and other | 36,979 | 72,521 | (35,542) | (49) | % | |||||||||||||||
| Total revenues | 156,931 | 184,705 | (27,774) | (15) | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | 70,565 | 61,888 | 8,677 | 14 | % | |||||||||||||||
| Selling, general and administrative | 50,476 | 57,589 | (7,113) | (12) | % | |||||||||||||||
| Research and development | 61,149 | 104,659 | (43,510) | (42) | % | |||||||||||||||
| Contingent consideration | (1,923) | (6,453) | 4,530 | (70) | % | |||||||||||||||
| Amortization of intangible assets | 20,183 | 22,382 | (2,199) | (10) | % | |||||||||||||||
| Asset impairment charges | — | 53,702 | (53,702) | (100) | % | |||||||||||||||
| Total costs and expenses | 200,450 | 293,767 | (93,317) | (32) | % | |||||||||||||||
| Loss from operations | (43,519) | (109,062) | 65,543 | (60) | % | |||||||||||||||
Revenue. The increase in revenue from products for the year ended December 31, 2020 compared to the year ended December 31, 2019 was primarily attributable to an increase in sales at OPKO Chile and an increase in sales of Rayaldee. Sales of Rayaldee were $36.8 million for the year ended December 31, 2020, as compared to $31.4 million for 2019. Revenue from transfer of intellectual property and other for the years ended December 31, 2020 and 2019 principally reflected $28.7 million and $66.8 million, respectively, of revenue related to the Pfizer Transaction. Revenue from transfer of intellectual property and other for the year ended December 31, 2020 also included a $3 million milestone payment triggered by the first marketing approval of Rayaldee in Europe.
Cost of revenue. Cost of revenue for the year ended December 31, 2020 increased $8.7 million compared to the year ended December 31, 2019. Cost of product revenue increased primarily due to an increase in sales at OPKO Chile and changes in product mix during the year ended December 31, 2020.
Selling, general and administrative expenses. Selling, general and administrative expenses for the years ended December 31, 2020 and 2019 were $50.5 million and $57.6 million, respectively. The decrease in selling, general and administrative expenses was primarily due to decreased expenses at our pharmaceutical subsidiaries and a decrease in equity-based compensation expense. Selling, general and administrative expenses for the pharmaceutical segment for the years ended December 31, 2020 and 2019 included equity-based compensation expense of $1.0 million and $2.0 million, respectively.
Research and development expenses. Research and development expenses for the years ended December 31, 2020 and 2019 were $61.1 million and $104.7 million, respectively. Research and development expenses include external and internal expenses, partially offset by third-party grants and funding arising from collaboration agreements. External expenses include clinical and non-clinical activities performed by contract research organizations, lab services, purchases of drug and diagnostic product materials and manufacturing development costs. We track external research and development expenses by individual program for phase 3 clinical trials for drug approval and premarket approval for diagnostics tests, if any. Internal expenses include employee-related expenses such as salaries, benefits and equity-based compensation expense. Other internal research and development expenses are incurred to support overall research and development activities and include expenses related to general overhead and facilities.
71
The following table summarizes the components of our research and development expenses:
| Research and Development Expenses | For the years ended December 31, | ||||||||||
| 2020 | 2019 | ||||||||||
| External expenses: | |||||||||||
| Manufacturing expense for biological products | $ | 5,326 | $ | 38,592 | |||||||
| Phase III studies | 10,513 | 16,869 | |||||||||
| Post-marketing studies | 1,270 | 2,019 | |||||||||
| Earlier-stage programs | 14,811 | 21,201 | |||||||||
| Research and development employee-related expenses | 21,931 | 22,023 | |||||||||
| Other internal research and development expenses | 9,440 | 8,813 | |||||||||
| Third-party grants and funding from collaboration agreements | (2,142) | (4,858) | |||||||||
| Total research and development expenses | $ | 61,149 | $ | 104,659 | |||||||
The decrease in research and development expenses for the year ended December 31, 2020 was primarily due to a decrease in research and development expenses related to Somatrogon, a once-weekly human growth hormone injection for which we have partnered with Pfizer and successfully completed a phase 3 study in August 2019. Ongoing expenses for the Somatrogon program support open label extension studies that will continue until the market launch of Somatrogon in certain countries, as well as the preparation of applications for marketing approvals. Research and development expenses for the pharmaceutical segment for the years ended December 31, 2020 and 2019 included equity-based compensation expense of $1.6 million and $2.1 million, respectively.
Contingent consideration. Contingent consideration for the years ended December 31, 2020 and 2019 was $1.9 million and $6.5 million reversal of expense, respectively. Contingent consideration for the years ended December 31, 2020 and 2019 was primarily attributable to changes in assumptions regarding the timing of achievement of future milestones for OPKO Renal, and potential amounts payable to former stockholders of OPKO Renal in connection therewith, pursuant to our acquisition agreement in March 2013.
Amortization of intangible assets. Amortization of intangible assets was $20.2 million and $22.4 million, respectively, for the years ended December 31, 2020 and 2019. Amortization expense reflects the amortization of acquired intangible assets with defined useful lives. Our indefinite lived IPR&D assets will not be amortized until the underlying development programs are completed. Upon obtaining regulatory approval by the U.S. FDA, the IPR&D assets will be accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life.
Asset impairment charges. Asset impairment charges were $53.7 million for the year ended December 31, 2019. Asset impairment charges for the year ended December 31, 2019 were primarily related to an impairment charge of $44.8 million to write our IPR&D assets for OPK88003 (oxyntomodulin) and CURNA’s platform technology for oligonucleotide therapeutics down to their estimated fair values, and a goodwill impairment charge of $8.2 million to write the carrying amount of the CURNA and Transition Therapeutics reporting units down to their estimated fair values. The Asset impairment charges for the year ended December 31, 2019, resulted from liquidity constraints, longer than expected development timelines and changes in the competitive landscape, which resulted in changes to our estimates and assumptions of the expected future cash flows associated with OPK88003 and CURNA’s platform technology.
We believe that our estimates and assumptions in testing goodwill and other intangible assets, including IPR&D, for impairment are consistent with assumptions that marketplace participants would use in their estimates. However, if actual results are not consistent with our estimates and assumptions, including as a result of the COVID-19 global pandemic, we may be exposed to an impairment charge that could be material. If we are unable to successfully develop Somatrogon, or changes in projections and assumptions negatively impact our forecast of net cash flows, we may be exposed to a material impairment charge related to the IPR&D for Somatrogon.
72
Corporate
| For the years ended December 31, | ||||||||||||||||||||
| (In thousands) | 2020 | 2019 | Change | % Change | ||||||||||||||||
| Revenues: | ||||||||||||||||||||
| Revenue from transfer of intellectual property and other | $ | — | $ | 796 | (796) | (100) | % | |||||||||||||
| Total revenues | — | 796 | (796) | (100) | % | |||||||||||||||
| Costs and expenses: | ||||||||||||||||||||
| Cost of revenue | (84) | (261) | 177 | (68) | % | |||||||||||||||
| Selling, general and administrative | 38,609 | 43,696 | (5,087) | (12) | % | |||||||||||||||
| Research and development | (836) | (1,008) | 172 | (17) | % | |||||||||||||||
| Total costs and expenses | 37,689 | 42,427 | (4,738) | (11) | % | |||||||||||||||
| Loss from operations | (37,689) | (41,631) | 3,942 | (9) | % | |||||||||||||||
Operating loss for our unallocated corporate operations for the years ended December 31, 2020 and 2019 was $37.7 million and $41.6 million, respectively, and principally reflects general and administrative expenses incurred in connection with our corporate operations. The decrease in operating loss for the year ended December 31, 2020 was primarily attributable to a decrease in legal fees incurred for the year ended December 31, 2020, as compared to the year ended December 31, 2019.
Other
Interest income. Interest income for the years ended December 31, 2020 and 2019 was not significant as our cash investment strategy emphasizes the security of the principal invested and fulfillment of liquidity needs.
Interest expense. Interest expense for the years ended December 31, 2020 and 2019 was $21.9 million and $21.5 million, respectively. Interest expense was principally related to interest incurred on our Senior Convertible Notes due 2025 (the “2025 Notes”), our 5% Convertible Promissory Notes (the “2023 Convertible Notes”), our 3.0% Senior Notes due 2033 (the “2033 Senior Notes”), and BioReference’s outstanding debt under its credit facility.
Fair value changes of derivative instruments, net. Fair value changes of derivative instruments, net for the years ended December 31, 2020 and 2019, was $50 thousand and $174 thousand of income, respectively. Derivative income for the year ended December 31, 2020, was principally related to the change in fair value on foreign currency forward exchange contracts at OPKO Chile.
Other income (expense), net. Other income (expense), net for the years ended December 31, 2020 and 2019, was $12.7 million of income and $11.3 million of expense, respectively. Other income for the year ended December 31, 2020 primarily consisted of realized and unrealized gains recognized during the period on our investment in VBI Vaccines Inc. (“VBI”), offset by net unrealized losses recognized during the period on our investment in Eloxx Pharmaceuticals, Inc. (“Eloxx”). Other expense for the year ended December 31, 2019 primarily consisted of net unrealized losses recognized during the period on Eloxx and VBI.
Income tax provision. Our income tax provision for the years ended December 31, 2020 and 2019 was $17.6 million and $7.1 million, respectively, and reflects results using our expected effective tax rate. For the year ended December 31, 2020, the tax rate differed from the U.S. federal statutory rate of 21% primarily due to the relative mix in earnings and losses in the U.S. versus foreign tax jurisdictions, the impact of certain discrete tax events and operating results in tax jurisdictions that do not result in a tax benefit.
Loss from investments in investees. We have made investments in certain early stage companies that we perceive to have valuable proprietary technology and significant potential to create value for us as a shareholder or member. We account for these investments under the equity method of accounting, resulting in the recording of our proportionate share of their losses until our share of their loss exceeds our investment. Until the investees’ technologies are commercialized, if ever, we anticipate they will report net losses. Loss from investments in investees was $0.5 million and $2.9 million for the years ended December 31, 2020 and 2019, respectively.
LIQUIDITY AND CAPITAL RESOURCES
At December 31, 2021, we had cash and cash equivalents of approximately $134.7 million. Cash provided by operations of $38.3 million for year ended December 31, 2021 principally reflects cash generated by our diagnostics segment due to the
73
positive impact of COVID-19 testing volumes, which was partially offset by general and administrative expenses related to our corporate operations and research and development activities. Cash provided by investing activities for the year ended December 31, 2021 primarily reflects $66.0 million from the sale of property, plant and equipment, which was partially offset by capital expenditures of $32.2 million. Cash used in financing activities of $10.4 million primarily reflects net repayments on our lines of credit. We have historically not generated sustained positive cash flow sufficient to offset our operating and other expenses, and our primary sources of cash have been from the public and private placement of equity, the issuance of the 2033 Senior Notes, 2023 Convertible Notes and 2025 Notes and credit facilities available to us. However, as a result of the significant increase in testing volumes resulting from the COVID-19 pandemic, we have generated positive cash flow from operations; however we are unable to predict how long the demand will continue for our COVID-19 related testing, or whether pricing and reimbursement policies for testing will sustain, and accordingly, the sustainability of our cash flows from operations. Overall demand for COVID-19 testing has recently declined, and accordingly, the sustainability of our COVID-19 testing volumes is uncertain.
In January 2022, we and Sema4 announced the execution of the GeneDx Merger Agreement, pursuant to which Sema4 has agreed to acquire our wholly owned subsidiary, GeneDx, Inc. The GeneDx Transaction is expected to close in the second quarter of 2022.
Under the terms of the GeneDx Merger Agreement, Sema4 has agreed to acquire GeneDx for an upfront payment of $150 million in cash, together with 80.0 million shares of Sema4 Common Stock, subject to a customary purchase price adjustment mechanism providing for a normalized level of working capital and that GeneDx be free of debt at closing of the GeneDx Transaction. Additionally, Sema4 agreed to pay OPKO up to an additional $150.0 million, which may be paid in Sema4 Common Stock, cash or a combination thereof in Sema4’s discretion, subject to GeneDx achieving certain revenue targets for the fiscal years ending December 31, 2022 and 2023. Based on the closing stock price of Sema4 Common Stock as of January 14, 2022, the total upfront consideration is approximately $473 million, and the total aggregate consideration including potential milestones is approximately $623 million.
As of December 31, 2021, GeneDx met the held-for-sale accounting criteria and the related assets and liabilities are classified as held for sale in the consolidated balance sheet. Depending upon the value Sema4 shares upon closing of the transaction, an impairment charge may be incurred. GeneDx was included in our diagnostics segment as of December 31, 2021.
In June 2021, EirGen Pharma Limited (“EirGen”), our wholly owned subsidiary, entered into a definitive agreement to sell one of its facilities in Waterford, Ireland to Horizon Therapeutics plc for $65 million in cash less certain assumed and accrued liabilities relating to transferred employees. The facility, which was formerly included in our pharmaceutical segment, housed EirGen’s sterile-fill-finish business and was no longer a core component of our ongoing operations and business strategy. The transaction closed in the third quarter of 2021.
On February 25, 2020, we entered into a credit agreement with an affiliate of Dr. Frost, pursuant to which the lender committed to provide us with an unsecured line of credit in the amount of $100 million. Borrowings under this line of credit incurred interest at a rate of 11% per annum and could be repaid and reborrowed at any time. The line of credit also called for a commitment fee equal to 0.25% per annum of the unused portion of the line. No funds were borrowed under this line of credit. We terminated this line of credit in June 2021.
In February 2019, we issued $200.0 million aggregate principal amount of the 2025 Notes in an underwritten public offering. The 2025 Notes bear interest at a rate of 4.50% per year, payable semiannually in arrears on February 15 and August 15 of each year. The notes mature on February 15, 2025, unless earlier repurchased, redeemed or converted.
Holders may convert their 2025 Notes into shares of Common Stock at their option at any time prior to the close of business on the business day immediately preceding November 15, 2024, subject to the satisfaction of certain conditions. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our Common Stock, or a combination of cash and shares of our Common Stock, at our election.
The current conversion rate for the 2025 Notes is 236.7424 shares of Common Stock per $1,000 principal amount of 2025 Notes (equivalent to a conversion price of approximately $4.22 per share of Common Stock). The conversion rate for the 2025 Notes is subject to adjustment in certain events but will not be adjusted for any accrued and unpaid interest.
In May 2021, we entered into exchange agreements with certain holders of the 2025 Notes pursuant to which the holders exchanged $55.4 million in aggregate principal amount of the outstanding 2025 Notes for 19,051,270 shares of our Common Stock (the “Exchange”). Upon consummation of the Exchange, we paid the holders of the exchanged notes an aggregate of approximately $0.6 million in accrued and unpaid interest on the exchanged notes. We recorded an $11.1 million non-cash loss related to the Exchange.
74
As of December 31, 2021, the total commitments under our A&R Credit Agreement (as defined below) with CB and our lines of credit with financial institutions in Chile and Spain were $90.2 million, of which $13.7 million was drawn as of December 31, 2021. At December 31, 2021, the weighted average interest rate on these lines of credit was approximately 5.4%. These lines of credit are short-term and are used primarily as a source of working capital. The highest aggregate principal balance at any time outstanding during the year ended December 31, 2021, was $66.5 million. We intend to continue to draw on these lines of credit as needed. There is no assurance that these lines of credit or other funding sources will be available to us on acceptable terms, or at all, in the future.
In November 2015, BioReference and certain of its subsidiaries entered into the Credit Agreement with CB, as lender, which was amended and restated on August 30, 2021 (the “A&R Credit Agreement”). The A&R Credit Agreement provides for a $75.0 million secured revolving credit facility and includes a $20.0 million sub-facility for swingline loans and a $20.0 million sub-facility for the issuance of letters of credit. The A&R Credit Agreement matures on August 30, 2024 and is guaranteed by all of BioReference’s domestic subsidiaries, subject to certain exceptions. The A&R Credit Agreement is also secured by substantially all assets of BioReference and its domestic subsidiaries, subject to certain exceptions, as well as a non-recourse pledge by us of our equity interest in BioReference. Availability under the A&R Credit Agreement is based on a borrowing base composed of eligible accounts receivables of BioReference and certain of its subsidiaries, as specified therein. As of December 31, 2021, $64.8 million remained available for borrowing under the A&R Credit Agreement.
In February 2018, in a transaction exempt from registration under the Securities Act, we issued the 2023 Convertible Notes in the aggregate principal amount of $55.0 million maturing in February 2023. Each holder of a 2023 Convertible Note has the option, from time to time, to convert all or any portion of the outstanding principal balance of such 2023 Convertible Note, together with accrued and unpaid interest thereon, into shares of our Common Stock, par value $0.01 per share, at a conversion price of $5.00 per share of Common Stock. We may redeem all or any part of the then issued and outstanding 2023 Convertible Notes, together with accrued and unpaid interest thereon upon no fewer than 30 days, and no more than 60 days, notice to the holders. The 2023 Convertible Notes contain customary events of default and representations and warranties of OPKO.
In December 2021, we announced that the FDA approved our 4Kscore test for use in men age 45 and older who have not had a prior prostate biopsy or are biopsy negative and have an age-specific abnormal total PSA and/or abnormal digital rectal exam.
In December 2021, we announced preliminary topline results from our Phase 2 trial with Rayaldee to treat mild-to-moderate COVID-19. This study builds on increasing medical evidence that vitamin D repletion therapy can mitigate the severity of upper respiratory tract infections and accelerate recovery from COVID-19.
One primary efficacy endpoint was reaching the targeted serum 25D level. By Day 7, mean serum 25D levels increased with Rayaldee treatment to 82 ng/mL (p<0.001) and remained elevated for the duration of the trial, with 88% of subjects attaining the targeted level. In contrast, mean 25D declined slightly with placebo treatment.
On September 14, 2021, we and LeaderMed Health Group Limited (“LeaderMed”), a pharmaceutical development company with operations based in Asia, announced the formation of a joint venture to develop, manufacture and commercialize two of OPKO’s clinical stage, long-acting drug products in Greater China and eight other Asian territories.
Under the terms of the agreements, we will grant the joint venture exclusive rights to develop, manufacture and commercialize (a) OPK88003, an oxyntomodulin analog being developed for the treatment of obesity and diabetes, and (b) Factor VIIa-CTP, a novel long-acting coagulation factor being developed to treat hemophilia, in exchange for a 47% ownership interest in the joint venture. In addition, we received an upfront payment of $1 million and will be reimbursed for clinical trial material and technical support it provides the joint venture.
LeaderMed will be responsible for funding the joint venture’s operations, development and commercialization efforts and will, with its syndicate partners, initially invest $11 million in exchange for a 53% ownership interest. We retain full rights to oxyntomodulin and Factor VIIa-CTP in all other geographies.
On August 16, 2021, BioReference announced the acquisition of Ariosa Diagnostics, Inc. (“Ariosa”), the U.S. Ariosa centralized laboratory prenatal testing business from Roche Molecular Systems Inc. Ariosa's non invasive prenatal screening (NIPS) test, the Harmony Prenatal Test, is one of the most widely studied tests utilized in prenatal screening. The test has been performed in over 1.5 million patients. BioReference currently offers, ClariTest™ Core, which utilizes the same core technology as the Harmony Prenatal Test. The acquisition of Ariosa will complement our current NIPS offering.
On July 6, 2021, we entered into the CAMP4 Agreement, pursuant to which we granted to CAMP4 an exclusive license to develop, manufacture, commercialize or improve therapeutics utilizing the AntagoNAT technology, an oligonucleotide
75
platform developed under OPKO CURNA, which includes the CAMP4 Licensed Compound and the CAMP4 Licensed Product, worldwide. The CAMP4 Agreement grant covers human pharmaceutical, prophylactic, and therapeutic and certain diagnostic uses.
We received an initial upfront payment of $1.5 million and 3,373,008 shares of CAMP4’s Preferred Stock, which equates to approximately 9% of the outstanding shares of CAMP4, and we are eligible to receive up to $3.5 million in development milestone payments for Dravet syndrome products, and $4 million for non-Dravet syndrome products, as well as sales milestones of up to $90 million for Dravet syndrome products and up to $90 million for non-Dravet syndrome products. We may also receive double digit royalty payments on the net sales of royalty bearing products, subject to adjustment. In addition, upon achievement of certain development milestones, we will be eligible to receive additional equity consideration of up to 5,782,299 shares of CAMP4 Preferred Stock in connection with Dravet syndrome products and up to 1,082,248 shares of CAMP4 Preferred Stock in connection with non-Dravet syndrome products.
Unless earlier terminated, the CAMP4 Agreement will remain in effect on a CAMP4 Licensed Product-by-Licensed Product and country by-country basis until such time as the royalty term expires for a CAMP4 Licensed Product in a country, and expires in its entirety upon the expiration of the royalty term for the last CAMP4 Licensed Product in the last country. CAMP4’s royalty obligations expire on the later of (i) the expiration, invalidation or abandonment date of the last patent right in connection with the royalty bearing product, or (ii) ten (10) years after a royalty bearing product’s first commercial sale in a country. In addition to termination rights for material breach and bankruptcy, CAMP4 is permitted to terminate the CAMP4 Agreement after a specified notice period.
On June 18, 2021, EirGen and Nicoya entered into a Development and License Agreement (the “Nicoya Agreement”) granting Nicoya the exclusive rights for the development and commercialization of extended release calcifediol (the “ Nicoya Product”) in Greater China, which includes mainland China, Hong Kong, Macau, and Taiwan (collectively, the “Nicoya Territory”). Extended release calcifediol is marketed in the U.S. under the tradename Rayaldee by OPKO. The license grant to Nicoya covers the therapeutic and preventative use of the Product for SHPT in non-dialysis (“ND”) and hemodialysis chronic kidney disease patients (the “Nicoya Field”).
EirGen received an initial upfront payment of $5 million and is eligible to receive an additional $5 million upon the first to occur of (A) a certain predetermined milestone, or (B) the first anniversary of the effective date. EirGen is also eligible to receive up to an additional aggregate amount of $115 million upon the achievement of certain development, regulatory and sales-based milestones by Nicoya for the Nicoya Product in the Nicoya Territory. EirGen will also receive tiered, double digit royalty payments at rates in the low double digits on net product sales within the Nicoya Territory and in the Nicoya Field.
In May 2016, EirGen partnered with VFMCRP through the VFMCRP Agreement for the development and commercialization of Rayaldee in the VFMCRP Territory. The license to VFMCRP potentially covers all therapeutic and prophylactic uses of the product in human patients, provided that initially the license is for the use of the product for the treatment or prevention of SHPT related to patients with CKD and vitamin D insufficiency/deficiency (“VFMCRP Initial Indication”). Effective May 23, 2021, we entered into an amendment to the VFMCRP Agreement, pursuant to which the parties thereto agreed to include Japan as part of the VFMCRP Territory. Effective May 5, 2020, we entered into the VFMCRP Amendment, pursuant to which the parties agreed to exclude Mexico, South Korea, the Middle East and all of the countries of Africa from the VFMCRP Territory. In addition, the parties agreed to certain amendments to the milestone structure and to reduce minimum royalties payable.
We have received non-refundable and non-creditable payments of $55 million to date and are eligible to receive up to an additional $227 million pursuant to the terms of the VFMCRP Amendment upon the achievement of certain regulatory and sales-based milestones tied to sales and reimbursement levels. In addition, we are eligible to receive tiered royalties on sales of the product at percentage rates that range from the mid-teens to the mid-twenties or a minimum royalty, whichever is greater, upon commencement of sales of the product.
As part of the arrangement, the companies will share responsibility for the conduct of trials specified within an agreed-upon development plan, with each company leading certain activities within the plan. For the initial development plan, the companies have agreed to certain cost sharing arrangements. VFMCRP will be responsible for all other development costs that VFMCRP considers necessary to develop the product for the VFMCRP Initial Indication in the VFMCRP Territory except as otherwise provided in the VFMCRP Agreement. EirGen also granted to VFMCRP an option (the “Option”) to acquire an exclusive license to use, import, offer for sale, sell, distribute and commercialize the product in the U.S. for treatment of SHPT in dialysis patients with stage 5 CKD and vitamin D insufficiency (the “Dialysis Indication”). Upon exercise of the Option, VFMCRP will reimburse EirGen for all of the development costs incurred by EirGen with respect to the product for the Dialysis Indication in the U.S. VFMCRP would also pay EirGen up to an additional aggregate amount of $555 million upon the achievement of certain milestones and would be obligated to pay royalties on sales of the product at percentage rates that
76
range from the mid-teens to the mid-twenties or a minimum royalty, whichever is greater, upon commencement of sales of the product.
In early 2022, the European Commission and Ministry of Health, Labour and Welfare in Japan approved the next-generation long-acting recombinant human growth hormone NGENLA (Somatrogon), a once-weekly injection to treat pediatric growth hormone deficiency. Further, Canada and Australia approved NGENLA in October and November of 2021, respectively.
In January 2022, the FDA issued a Complete Response Letter for the BLA for Somatrogon. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States.
In 2014, Pfizer and OPKO entered into a worldwide agreement for the development and commercialization of our long-acting Somatrogon for the treatment of GHD in adults and children, as well as for the treatment of growth failure in children born small for gestational age. In May 2020, we entered into the Restated Pfizer Agreement which was effective as of January 1, 2020, pursuant to which the parties agreed to share all costs for Manufacturing Activities, as defined in the Restated Pfizer Agreement, for developing a licensed product for the three indications included in the Restated Pfizer Agreement. Under the terms of the agreements with Pfizer, we received non-refundable and non-creditable upfront payments of $295 million in 2015 and are eligible to receive up to an additional $275 million upon the achievement of certain regulatory milestones. Pfizer received the exclusive license to commercialize Somatrogon worldwide. In addition, we are eligible to receive initial tiered royalty payments associated with the commercialization of Somatrogon for Adult GHD with percentage rates ranging from the high teens to mid-twenties. Upon the launch of Somatrogon for Pediatric GHD in certain major markets, the royalties will transition to regional, tiered gross profit sharing for both Somatrogon and Pfizer’s Genotropin®. During the first quarter of 2021, regulatory submissions in the major global markets for Somatrogan have been accepted including, the U.S., European Medicines Agency, and Ministry of Health, Labour, and Welfare in Japan for Somatrogon for the treatment of pediatric patients with GHD.
In early 2022, the European Commission and Ministry of Health, Labour and Welfare in Japan approved the next-generation long-acting recombinant human growth hormone NGENLA (Somatrogon), a once-weekly injection to treat pediatric growth hormone deficiency. Further, Canada and Australia approved NGENLA in October and November of 2021, respectively.
In January 2022, the FDA issued a Complete Response Letter for the BLA for Somatrogon. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States.
In connection with our acquisitions of CURNA, OPKO Diagnostics and OPKO Renal, we agreed to pay future consideration to the sellers upon the achievement of certain events, including up to an additional $19.1 million in shares of our Common Stock to the former stockholders of OPKO Diagnostics upon and subject to the achievement of certain milestones; and up to an additional $125.0 million in either shares of our Common Stock or cash, at our option subject to the achievement of certain milestones, to the former shareholders of OPKO Renal. As a result of our execution of the CAMP4 Agreement, we will have to pay a percentage of any payments received under the CAMP4 Agreement to the former CURNA stockholders.
We believe that the cash and cash equivalents on hand at December 31, 2021, cash from operations and the amounts available to be borrowed under our lines of credit are sufficient to meet our anticipated cash requirements for operations and debt service beyond the next 12 months. We based this estimate on assumptions that may prove to be wrong or are subject to change, and we may be required to use our available cash resources sooner than we currently expect. If we acquire additional assets or companies, accelerate our product development programs or initiate additional clinical trials, we will need additional funds. Our future cash requirements, and the timing of those requirements, will depend on a number of factors, including the evolving impact of the COVID-19 pandemic on our business, the approval and success of our products in development, particularly our long acting Somatrogon for which we have received approval in Europe, Japan, Australia and Canada, submitted for approval in the U.S. and received a Complete Response Letter in January 2022, the approval and success of Somatrogon outside the United States, including in Europe, Japan, Australia and Canada, the commercial success of Rayaldee, including from the recent launch of Rayaldee by Vifor and in other territories expected in 2022, BioReference’s financial performance, possible acquisitions and dispositions, the continued progress of research and development of our product candidates, the timing and outcome of clinical trials and regulatory approvals, the costs involved in preparing, filing, prosecuting, maintaining, defending, and enforcing patent claims and other intellectual property rights, the status of competitive products, the availability of financing, our success in developing markets for our product candidates and results of government investigations, payor claims, and legal proceedings that may arise, including, without limitation class action and derivative litigation to which we are subject, and our ability to obtain insurance coverage for such claims. We have historically not
77
generated sustained positive cash flow and if we are not able to secure additional funding when needed, we may have to delay, reduce the scope of, or eliminate one or more of our clinical trials or research and development programs or possible acquisitions or reduce our marketing or sales efforts or cease operations.
Additionally, the rapid development and fluidity of the COVID-19 pandemic and new variants of the virus makes it very difficult to predict its ultimate impact on our business, results of operations and liquidity. The pandemic presents a significant uncertainty that could materially and adversely affect our results of operations, financial condition and cash flows, including a negative impact on non-COVID-related diagnostics testing services provided by BioReference in our diagnostics segment, notwithstanding that our results of operations have been positively impacted by our provision of COVID-19 testing services. Further, deteriorating economic conditions globally as a result of the COVID-19 pandemic have in the past resulted, and may in the future result in a challenging capital raising environment, which could materially limit our access to capital, whether through the issuance and sale of our Common Stock, debt securities or otherwise, as well as through bank facilities and lines of credit. Events resulting from the effects of COVID-19 or new variants of the virus could negatively impact our ability to comply with certain covenants in the A&R Credit Agreement or require that we pursue alternative financing. We can provide no assurance that any such alternative financing, if required, could be obtained on acceptable terms or at all. The combination of potential disruptions to our business resulting from COVID-19 together with and volatile credit and capital markets could adversely impact our future liquidity, which could have an adverse effect on our business and results of operations. We will continue to monitor and assess the impact COVID-19 and new variants of the virus may have on our business and financial results.
The following table provides information as of December 31, 2021, with respect to the amounts and timing of our known contractual obligation payments due by period.
| Contractual obligations (In thousands) |
2022 | 2023 | 2024 | 2025 | 2026 | Thereafter | Total | |||||||||||||||||||||||||||||||||||||
| Open purchase orders | $ | 249,279 | $ | 5,841 | $ | — | $ | — | $ | — | $ | — | $ | 255,120 | ||||||||||||||||||||||||||||||
| Operating leases | 11,624 | 8,770 | 5,693 | 3,599 | 3,043 | 11,992 | 44,721 | |||||||||||||||||||||||||||||||||||||
| Capital leases | 2,102 | 1,499 | 1,002 | 499 | 79 | — | 5,181 | |||||||||||||||||||||||||||||||||||||
| 2033 Senior Notes, 2025 and 2023 Convertible Notes | — | 68,576 | — | 119,360 | — | — | 187,935 | |||||||||||||||||||||||||||||||||||||
| Deferred payments | 2,478 | — | — | — | — | — | 2,478 | |||||||||||||||||||||||||||||||||||||
| Mortgages and other debts payable | 1,230 | 970 | 883 | 512 | 277 | — | 3,872 | |||||||||||||||||||||||||||||||||||||
| Lines of credit | 13,672 | — | — | — | — | — | 13,672 | |||||||||||||||||||||||||||||||||||||
| Interest commitments | 9,426 | 6,969 | 6,536 | 575 | 1 | — | 23,507 | |||||||||||||||||||||||||||||||||||||
| Total | $ | 289,811 | $ | 92,625 | $ | 14,114 | $ | 124,545 | $ | 3,400 | $ | 11,992 | $ | 536,486 | ||||||||||||||||||||||||||||||
The preceding table does not include information where the amounts of the obligations are not currently determinable, including the following:
•Contractual obligations in connection with clinical trials, which span over two years, and that depend on patient enrollment. The total amount of expenditures is dependent on the actual number of patients enrolled and as such, the contracts do not specify the maximum amount we may owe.
•Product license agreements effective during the lesser of 15 years or patent expiration whereby payments and amounts are determined by applying a royalty rate on uncapped future sales.
•Contingent consideration that includes payments upon achievement of certain milestones including meeting development milestones such as the completion of successful clinical trials, NDA approvals by the FDA and revenue milestones upon the achievement of certain revenue targets all of which are anticipated to be paid within the next seven years and are payable in either shares of our Common Stock or cash, at our option, and that may aggregate up to $144.1 million.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Use of estimates. The preparation of financial statements in conformity with accounting principles generally accepted in the U.S. (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ significantly from these estimates.
78
Goodwill and intangible assets. Goodwill and other intangible assets, including IPR&D, acquired in business combinations, licensing and other transactions at December 31, 2021 and 2020, was $1.4 billion and $1.7 billion, respectively.
Assets acquired and liabilities assumed in business combinations, licensing and other transactions are generally recognized at the date of acquisition at their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recognized as goodwill. At acquisition, we generally determine the fair value of intangible assets, including IPR&D, using the “income method.” This method starts with a forecast of net cash flows, risk adjusted for estimated probabilities of technical and regulatory success (for IPR&D) and adjusted to present value using an appropriate discount rate that reflects the risk associated with the cash flow streams.
Subsequent to acquisition, goodwill and indefinite lived intangible assets are tested at least annually as of October 1 for impairment, or when events or changes in circumstances indicate it is more likely than not that the carrying amount of such assets may not be recoverable. Our annual assessment may consist of a qualitative or quantitative analysis to determine whether it is more likely than not that its fair value exceeds the carrying value. When performing qualitative analysis, the factors we consider include our share price, our financial performance compared to budgets, long-term financial plans, the timing and cost of development plans, macroeconomic, industry and market conditions as well as the excess of fair value over the carrying value of net assets from the annual impairment test previously performed.
When performing quantitative analysis, we use a combination of income and market valuation methods and may weigh the outcomes of valuation approaches when estimating fair value. Inputs and assumptions used to determine fair value are determined from a market participant view, which might be different than our specific views. The valuation process is complex and requires significant input and judgment using internal and external sources. Market approaches depend on the availability of guideline companies and representative transactions. When using the income approach, complex and judgmental matters applicable to the valuation process may include the following:
•Estimated useful life – The asset life expected to contribute meaningful cash flows is determined after considering expected regulatory approval dates (if unapproved), exclusivity periods and other legal, regulatory or contractual provisions.
•Projections – Future revenues are estimated after considering many factors such as historical results, market opportunity, pricing, sales trajectories to peak sales levels, competitive environment and product evolution. Future costs and expenses are estimated after considering historical factors such as the timing and level of development costs to obtain regulatory approvals, maintain or further enhance the product. For IPR&D projects, we generally assume initial positive cash flows to commence shortly after the receipt of expected regulatory approvals which typically may not occur for a number of years. Actual cash flows attributed to the project are likely to be different than those assumed since projections are subjected to multiple factors including trial results and regulatory matters which could materially change the ultimate commercial success of the asset as well as significantly alter the costs to develop the respective asset into commercially viable products.
•Tax rates – The expected future income is tax effected using a market participant tax rate. In determining the tax rate, we consider the jurisdiction in which the intellectual property is held and the location of the research and manufacturing infrastructure.
•Discount rate – Discount rates are selected after considering the risks inherent in the future cash flows; the assessment of the asset’s life cycle and the competitive trends impacting the asset, including consideration of any Company specific technical, legal, regulatory, or economic barriers to entry.
Goodwill was $520.6 million and $680.6 million, respectively, at December 31, 2021 and 2020. In addition, at December 31, 2021, Assets held for sale includes $151.8 million of goodwill related to GeneDx. Estimating the fair value of a reporting unit for goodwill impairment is highly sensitive to changes in projections and assumptions and changes in assumptions could potentially lead to impairment. We perform sensitivity analyses around our assumptions in order to assess the reasonableness of the assumptions and the results of our testing. Ultimately, potential changes in these assumptions may impact the estimated fair value of a reporting unit and result in an impairment if the fair value of such reporting unit is less than its carrying value.
Net intangible assets other than goodwill were $911.9 million and $1.1 billion, including IPR&D of $590.2 million, at December 31, 2021 and December 31, 2020. Intangible assets are highly vulnerable to impairment charges, particularly newly acquired assets for recently launched products and IPR&D. Considering the high risk nature of research and development and the industry’s success rate of bringing developmental compounds to market, IPR&D impairment charges may occur in future periods. Estimating the fair value of IPR&D for potential impairment is highly sensitive to changes in projections and assumptions and changes in assumptions could potentially lead to impairment.
79
Upon obtaining regulatory approval, IPR&D assets are then accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life. If the project is abandoned, the IPR&D asset is charged to expense. Finite lived intangible assets are tested for impairment when events or changes in circumstances indicate it is more likely than not that the carrying amount of such assets may not be recoverable. The testing includes a comparison of the carrying amount of the asset to its estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the asset.
No impairment charges were recognized for the year ended December 31. 2021. and December 31, 2020. Impairment charges for the year ended December 31, 2019 were $92.4 million and consist of a goodwill impairment charge of $26.2 million to write the carrying amount of the OPKO Diagnostics, CURNA and Transition Therapeutics reporting units down to their estimated fair value, an impairment charge of $44.8 million to write our IPR&D assets for OPK88003 and CURNA’s platform technology for oligonucleotide therapeutics down to their estimated fair value, and an impairment charge of $20.7 million to write our intangible asset for the Claros Analyzer down to its estimated fair value as a result of our testing. These impairment charges for the year ended December 31, 2019, resulted from liquidity constraints, longer than expected development timelines and changes in the competitive landscape, which resulted in changes to our estimates and assumptions of the expected future cash flows of the reporting units focused on the development of the Claros Analyzer, OPK88003 and CURNA’s platform technology.
We believe that our estimates and assumptions in testing goodwill and other intangible assets, including IPR&D, for
impairment are reasonable and otherwise consistent with assumptions that marketplace participants would use in their estimates of fair value. However, if future results are not consistent with our estimates and assumptions, including as a result of the COVID-19 global pandemic, then we may be exposed to additional impairment charges, which could be material. Our 2021 impairment test of the OPKO Biologics reporting unit, including IPR&D related to Somatrogon, indicated an excess of estimated fair value over the carrying amount of approximately 19%. We submitted the initial BLA with the FDA for approval of Somatrogon (hGH-CTP) in the United States and Pfizer received a Complete Response Letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon in the United States. If we are unable to successfully commercialize Somatrogon in the U.S., or changes in projections and assumptions negatively impact our forecast of net cash flows, we may be exposed to a material impairment charge related to the IPR&D for Somatrogon.
We amortize intangible assets with definite lives on a straight-line basis over their estimated useful lives, ranging from 3 to 20 years. We use the straight-line method of amortization as there is no reliably determinable pattern in which the economic benefits of our intangible assets are consumed or otherwise used up. Amortization expense was $50.3 million, $56.4 million and $64.8 million for the years ended December 31, 2021, 2020 and 2019, respectively.
Revenue recognition. We generate revenues from services, products and intellectual property as follows:
Revenue from services. Revenue for laboratory services is recognized at the time test results are reported, which approximates when services are provided and the performance obligations are satisfied. Services are provided to patients covered by various third-party payor programs including various managed care organizations, as well as the Medicare and Medicaid programs. Billings for services are included in revenue net of allowances for contractual discounts, allowances for differences between the amounts billed and estimated program payment amounts, and implicit price concessions provided to uninsured patients which are all elements of variable consideration.
The following are descriptions of our payors for laboratory services:
Healthcare Insurers. Reimbursements from healthcare insurers are based on negotiated fee-for-service schedules. Revenues consist of amounts billed, net of contractual allowances for differences between amounts billed and the estimated consideration we expect to receive from such payors, which considers historical denial and collection experience and the terms of our contractual arrangements. Adjustments to the allowances, based on actual receipts from the third-party payors, are recorded upon settlement.
Government Payors. Reimbursements from government payors are based on fee-for-service schedules set by governmental authorities, including traditional Medicare and Medicaid. Revenues consist of amounts billed, net of contractual allowances for differences between amounts billed and the estimated consideration we expect to receive from such payors, which considers historical denial and collection experience and the terms of our contractual arrangements. Adjustments to the allowances, based on actual receipts from the government payors, are recorded upon settlement.
80
Client Payors. Client payors include physicians, hospitals, employers, and other institutions for which services are performed on a wholesale basis, and are billed and recognized as revenue based on negotiated fee schedules. Client payors also
include cities, states and companies for which BioReference provides COVID-19 testing services.
Patients. Uninsured patients are billed based on established patient fee schedules or fees negotiated with physicians on behalf of their patients. Insured patients (including amounts for coinsurance and deductible responsibilities) are billed based on fees negotiated with healthcare insurers. Collection of billings from patients is subject to credit risk and ability of the patients to pay. Revenues consist of amounts billed net of discounts provided to uninsured patients in accordance with our policies and implicit price concessions. Implicit price concessions represent differences between amounts billed and the estimated consideration that we expect to receive from patients, which considers historical collection experience and other factors including current market conditions. Adjustments to the estimated allowances, based on actual receipts from the patients, are recorded upon settlement.
The complexities and ambiguities of billing, reimbursement regulations and claims processing, as well as considerations unique to Medicare and Medicaid programs, require us to estimate the potential for retroactive adjustments as an element of variable consideration in the recognition of revenue in the period the related services are rendered. Actual amounts are adjusted in the period those adjustments become known. For the years ended December 31, 2021, and December 31, 2020, positive revenue adjustments due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $40.4 million and $0.3 million were recognized, respectively. For the year ended December 31, 2019, revenue reductions due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $24.8 million were recognized.
Third-party payors, including government programs, may decide to deny payment or recoup payments for testing they contend were improperly billed or not medically necessary, against their coverage determinations, or for which they believe they have otherwise overpaid (including as a result of their own error), and we may be required to refund payments already received. Our revenues may be subject to retroactive adjustment as a result of these factors among others, including without limitation, differing interpretations of billing and coding guidance and changes by government agencies and payors in interpretations, requirements, and “conditions of participation” in various programs. We have processed requests for recoupment from third-party payors in the ordinary course of our business, and it is likely that we will continue to do so in the future. If a third-party payor denies payment for testing or recoups money from us in a later period, reimbursement for our testing could decline.
As an integral part of our billing compliance program, we periodically assess our billing and coding practices, respond to payor audits on a routine basis, and investigate reported failures or suspected failures to comply with federal and state healthcare reimbursement requirements, as well as overpayment claims which may arise from time to time without fault on the part of the Company. We may have an obligation to reimburse Medicare, Medicaid, and third-party payors for overpayments regardless of fault. We have periodically identified and reported overpayments, reimbursed payors for overpayments and taken appropriate corrective action.
Settlements with third-party payors for retroactive adjustments due to audits, reviews or investigations are also considered variable consideration and are included in the determination of the estimated transaction price for providing services. These settlements are estimated based on the terms of the payment agreement with the payor, correspondence from the payor and our historical settlement activity, including an assessment of the probability a significant reversal of cumulative revenue recognized will occur when the uncertainty is subsequently resolved. Estimated settlements are adjusted in future periods as adjustments become known (that is, new information becomes available), or as years are settled or are no longer subject to such audits, reviews, and investigations. As of December 31, 2021 and 2020, we have liabilities of approximately $5.0 million and $14.9 million within Accrued expenses and Other long-term liabilities related to reimbursements for payor overpayments.
Revenue from products. We recognize revenue from product sales when a customer obtains control of promised goods or services. The amount of revenue that is recorded reflects the consideration that we expect to receive in exchange for those goods or services. Our estimates for sales returns and allowances are based upon the historical patterns of product returns and allowances taken, matched against the sales from which they originated, and our evaluation of specific factors that may increase or decrease the risk of product returns. Product revenues are recorded net of estimated rebates, chargebacks, discounts, co-pay assistance and other deductions (collectively, “Sales Deductions”) as well as estimated product returns which are all elements of variable consideration. Allowances are recorded as a reduction of revenue at the time product revenues are recognized. The actual amounts of consideration ultimately received may differ from our estimates. If actual results in the future vary from our estimates, we will adjust these estimates, which would affect Revenue from products in the period such variances become known.
81
Rayaldee is distributed in the U.S. principally through the retail pharmacy channel, which initiates with the largest wholesalers in the U.S. (collectively, “Rayaldee Customers”). In addition to distribution agreements with Rayaldee Customers, we have entered into arrangements with many healthcare providers and payors that provide for government-mandated and/or privately-negotiated rebates, chargebacks and discounts with respect to the purchase of Rayaldee.
We recognize revenue for shipments of Rayaldee at the time of delivery to customers after estimating Sales Deductions and product returns as elements of variable consideration utilizing historical information and market research projections. For the years ended December 31, 2021, 2020 and 2019, we recognized $27.0 million, $36.8 million and $31.4 million in net product revenue from sales of Rayaldee.
Taxes collected from customers related to revenues from services and revenues from products are excluded from revenues.
Revenue from intellectual property. We recognize revenues from the transfer of intellectual property generated through license, development, collaboration and/or commercialization agreements. The terms of these agreements typically include payment to us for one or more of the following: non-refundable, up-front license fees; development and commercialization milestone payments; funding of research and/or development activities; and royalties on sales of licensed products. Revenue is recognized upon satisfaction of a performance obligation by transferring control of a good or service to the customer.
For research, development and/or commercialization agreements that result in revenues, we identify all material performance obligations, which may include a license to intellectual property and know-how, and research and development activities. In order to determine the transaction price, in addition to any upfront payment, we estimate the amount of variable consideration at the outset of the contract either utilizing the expected value or most likely amount method, depending on the facts and circumstances relative to the contract. We constrain (reduce) our estimates of variable consideration such that it is probable that a significant reversal of previously recognized revenue will not occur throughout the life of the contract. When determining if variable consideration should be constrained, we consider whether there are factors outside of our control that could result in a significant reversal of revenue. In making these assessments, we consider the likelihood and magnitude of a potential reversal of revenue. These estimates are re-assessed each reporting period as required.
Upfront License Fees: If a license to our intellectual property is determined to be functional intellectual property distinct from the other performance obligations identified in the arrangement, we recognize revenue from nonrefundable, upfront license fees based on the relative value prescribed to the license compared to the total value of the arrangement. The revenue is recognized when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are not distinct from other obligations identified in the arrangement, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If the combined performance obligation is satisfied over time, we apply an appropriate method of measuring progress for purposes of recognizing revenue from nonrefundable, upfront license fees. We evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition.
Development and Regulatory Milestone Payments: Depending on facts and circumstances, we may conclude that it is appropriate to include the milestone in the estimated transaction price or that it is appropriate to fully constrain the milestone. A milestone payment is included in the transaction price in the reporting period that we conclude that it is probable that recording revenue in the period will not result in a significant reversal in amounts recognized in future periods. We may record revenues from certain milestones in a reporting period before the milestone is achieved if we conclude that achievement of the milestone is probable and that recognition of revenue related to the milestone will not result in a significant reversal in amounts recognized in future periods. We record a corresponding contract asset when this conclusion is reached. Milestone payments that have been fully constrained are not included in the transaction price to date. These milestones remain fully constrained until we conclude that achievement of the milestone is probable and that recognition of revenue related to the milestone will not result in a significant reversal in amounts recognized in future periods. We re-evaluate the probability of achievement of such development milestones and any related constraint each reporting period. We adjust our estimate of the overall transaction price, including the amount of revenue recorded, if necessary.
Research and Development Activities: If we are entitled to reimbursement from our customers for specified research and development expenses, we account for them as separate performance obligations if distinct. We also determine whether the research and development funding would result in revenues or an offset to research and development expenses in accordance with provisions of gross or net revenue presentation. The corresponding revenues or offset to research and development expenses are recognized as the related performance obligations are satisfied.
Sales-based Milestone and Royalty Payments: Our customers may be required to pay us sales-based milestone payments or royalties on future sales of commercial products. We recognize revenues related to sales-based milestone and royalty payments upon the later to occur of (i) achievement of the customer’s underlying sales or (ii) satisfaction of any performance
82
obligation(s) related to these sales, in each case assuming the license to our intellectual property is deemed to be the predominant item to which the sales-based milestones and/or royalties relate.
Other Potential Products and Services: Arrangements may include an option for license rights, future supply of drug substance or drug product for either clinical development or commercial supply at the licensee’s election. We assess if these options provide a material right to the licensee and if so, they are accounted for as separate performance obligations at the inception of the contract and revenue is recognized only if the option is exercised and products or services are subsequently delivered or when the rights expire. If the promise is based on market terms and not considered a material right, the option is accounted for if and when exercised. If we are entitled to additional payments when the licensee exercises these options, any additional payments are generally recorded in license or other revenues when the licensee obtains control of the goods, which is upon delivery.
For the years ended December 31, 2021, 2020 and 2019 we recorded $25.8 million, $53.2 million and $73.3 million of revenue from the transfer of intellectual property and other, respectively. For the year ended December 31, 2021, revenue from transfer of intellectual property and other principally reflects $10.8 million of revenue related to the Pfizer Transaction, $1.0 million related to the LeaderMed joint venture (as defined below), a $4.9 million payment received under the CAMP4 Agreement (as defined below) and a $5.0 million non-refundable upfront payment received under the Nicoya Agreement. For the years ended December 31, 2020, and 2019 revenue from transfer of intellectual property and other principally reflects $28.7 million and $66.8 million of revenue related to the Pfizer Transaction. In addition, revenue from the transfer of intellectual property and other for the year ended December 31, 2020 included $16.2 million of grants received by BioReference under the CARES Act and a $3 million milestone payment triggered by the first marketing approval of Rayaldee in Europe.
Contract liabilities relate to cash consideration that OPKO receives in advance of satisfying the related performance obligations. Changes in the contractual liabilities balance for the years ended December 31, 2021 are as follows:
| (In thousands) | ||||||||
| Balance at December 31, 2020 | $ | 16,378 | ||||||
| Balance at December 31, 2021 | 466 | |||||||
| Revenue recognized in the period from: | ||||||||
| Amounts included in contracts liability at the beginning of the period | $ | 15,911 | ||||||
The contract liability balance at December 31, 2020 related primarily to accelerated payments received as part of the CARES Act.
Concentration of credit risk and allowance for doubtful accounts. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of accounts receivable. Substantially all of our accounts receivable are with either companies in the health care industry or patients. However, credit risk is limited due to the number of our clients as well as their dispersion across many different geographic regions.
While we have receivables due from federal and state governmental agencies, we do not believe that such receivables represent a credit risk since the related healthcare programs are funded by federal and state governments, and payment is primarily dependent upon submitting appropriate documentation. At December 31, 2021 and 2020, receivable balances (net of explicit and implicit price concessions) from Medicare and Medicaid were 8% and 6%, respectively, of our consolidated Accounts receivable, net. At December 31, 2021, receivable balances (net of explicit and implicit price concessions) due directly from states, cities and other municipalities, specifically related to our real-time reverse-transcription polymerase chain reaction (real-time RT-PCR) assay to detect COVID-19, were 4.1% of our consolidated accounts receivable, net.
The portion of our accounts receivable due from individual patients comprises the largest portion of credit risk. At December 31, 2021 and 2020, receivables due from patients represented approximately 1.7% and 0.7%, respectively, of our consolidated accounts receivable, net.
We assess the collectability of accounts receivable balances by considering factors such as historical collection experience, customer credit worthiness, the age of accounts receivable balances, regulatory changes and current economic conditions and trends that may affect a customer’s ability to pay. Actual results could differ from those estimates. The allowance for credit losses was $1.8 million and $2.1 million at December 31, 2021 and 2020, respectively. The credit loss expense for the years ended December 31, 2021, 2020 and 2019 was $0.4 million, $0.2 million and $0.5 million, respectively.
83
Income taxes. Income taxes are accounted for under the asset-and-liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and the respective tax bases and for operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. We periodically evaluate the realizability of our net deferred tax assets. Our tax accruals are analyzed periodically and adjustments are made as events occur to warrant such adjustment. Valuation allowances on certain U.S. deferred tax assets and non-U.S. deferred tax assets are established, because realization of these tax benefits through future taxable income does not meet the more-likely-than-not threshold.
Equity-based compensation. We measure the cost of services received in exchange for an award of equity instruments based on the grant-date fair value of the award. That cost is recognized in the Consolidated Statements of Operations over the period during which an employee is required to provide service in exchange for the award. We record excess tax benefits, realized from the exercise of stock options, as cash flows from operations. We estimate the grant-date fair value of our stock option grants using a valuation model known as the Black-Scholes-Merton formula or the “Black-Scholes Model.” The Black-Scholes Model requires the use of several variables to estimate the grant-date fair value of stock options including expected term, expected volatility, expected dividends and risk-free interest rate. We perform analyses to calculate and select the appropriate variable assumptions used in the Black-Scholes Model. The selection of assumptions is subject to significant judgment and future changes to our assumptions and estimates which may have a material impact on our Consolidated Financial Statements.
Inventories. Inventories are valued at the lower of cost and net realizable value. Cost is determined by the first-in, first-out method. We consider such factors as the amount of inventory on hand, estimated time required to sell such inventories, remaining shelf-life, and current market conditions to determine whether inventories are stated at the lower of cost and net realizable value. Inventories at our diagnostics segment consist primarily of purchased laboratory supplies, which are used in our testing laboratories. Inventory obsolescence expense for the years ended December 31, 2021, 2020 and 2019 was $6.5 million, $4.4 million and $2.3 million, respectively.
Contingent consideration. Each period we revalue the contingent consideration obligations associated with certain prior acquisitions to their fair value and record increases in the fair value as contingent consideration expense and decreases in the fair value as a reduction in contingent consideration expense. Changes in contingent consideration result from changes in the assumptions regarding probabilities of successful achievement of related milestones, the estimated timing in which the milestones are achieved and the discount rate used to estimate the fair value of the liability. Contingent consideration may change significantly as our development programs progress, revenue estimates evolve and additional data is obtained, impacting our assumptions. The assumptions used in estimating fair value require significant judgment. The use of different assumptions and judgments could result in a materially different estimate of fair value which may have a material impact on our results from operations and financial position.
RECENT ACCOUNTING PRONOUNCEMENTS
Pending accounting pronouncements.
In August 2020, the FASB issued ASU No. 2020-06, “Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity's Own Equity (Subtopic 815-40).” ASU 2020-06 will simplify the accounting for convertible instruments by reducing the number of accounting models for convertible debt instruments and convertible preferred stock. The ASU is effective for public entities for fiscal years beginning after December 15, 2021, with early adoption permitted. We are currently evaluating the impact of this new guidance on our Consolidated Financial Statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
In the normal course of doing business, we are exposed to the risks associated with foreign currency exchange rates and changes in interest rates.
Foreign Currency Exchange Rate Risk – We operate globally and, as such, we are subject to foreign exchange risk in our commercial operations as portions of our revenues are exposed to changes in foreign currency exchange rates, primarily the Chilean Peso, the Mexican Peso, and the Euro.
Although we do not speculate in the foreign exchange market, we may from time to time manage exposures that arise in the normal course of business related to fluctuations in foreign currency exchange rates by entering into offsetting positions through the use of foreign exchange forward contracts. Certain firmly committed transactions may be hedged with foreign
84
exchange forward contracts. As exchange rates change, gains and losses on the exposed transactions are partially offset by gains and losses related to the hedging contracts. Both the exposed transactions and the hedging contracts are translated and fair valued, respectively, at current spot rates, with gains and losses included in earnings.
Our derivative activities, which consist of foreign exchange forward contracts, are initiated to economically hedge forecasted cash flows that are exposed to foreign currency risk. The foreign exchange forward contracts generally require us to exchange local currencies for foreign currencies based on pre-established exchange rates at the contracts’ respective maturity dates. As exchange rates change, gains and losses on these contracts are generated based on the change in the exchange rates that are recognized in the Consolidated Statements of Operations and offset the impact of the change in exchange rates on the foreign currency cash flows that are hedged. If the counterparties to the exchange contracts do not fulfill their obligations to deliver the contracted currencies, we could be at risk for currency related fluctuations. Our foreign exchange forward contracts primarily hedge exchange rates on the Chilean Peso to the U.S. dollar. If Chilean Pesos were to strengthen or weaken in relation to the U.S. dollar, our loss or gain on hedged foreign currency cash-flows would be offset by the derivative contracts, with a net effect of zero.
We do not engage in trading market risk sensitive instruments or purchasing hedging instruments or “other than trading” instruments that are likely to expose us to significant market risk, whether interest rate, foreign currency exchange, commodity price, or equity price risk.
Interest Rate Risk – Our exposure to interest rate risk relates to our cash and investments and to our borrowings. We generally maintain an investment portfolio of money market funds and marketable securities. The securities in our investment portfolio are not leveraged, and are, due to their very short-term nature, subject to minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that a change in market interest rates would have a significant negative impact on the value of our investment portfolio except for reduced income in a low interest rate environment.
At December 31, 2021, we had cash and cash equivalents of $134.7 million. The weighted average interest rate related to our cash and cash equivalents for the year ended December 31, 2021 was less than 1%. As of December 31, 2021, there was no principal outstanding balance under the A&R Credit Agreement with CB and the principal outstanding balance under our Chilean and Spanish lines of credit was $13.7 million in the aggregate at a weighted average interest rate of approximately 5.4%.
Our $3.0 million aggregate principal amount of our 2033 Senior Notes has a fixed interest rate of 3%, our $55.0 million aggregate principal amount of our 2023 Convertible Notes has a fixed interest rate of 5%, and our $200.0 million aggregate principal amount of the 2025 Notes has a fixed interest rate of 4.50%, and therefore are not subject to fluctuations in market interest rates.
The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we may invest our excess cash in debt instruments of the U.S. Government and its agencies, bank obligations, repurchase agreements and high-quality corporate issuers, and money market funds that invest in such debt instruments, and, by policy, restrict our exposure to any single corporate issuer by imposing concentration limits. To minimize the exposure due to adverse shifts in interest rates, we maintain investments at an average maturity of generally less than three months.
85
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
| Page | |||||
Reports of Independent Registered Certified Public Accounting Firm (PCAOB ID No. |
|||||
Consolidated Balance Sheets |
|||||
Consolidated Statements of Operations |
|||||
Consolidated Statements of Comprehensive Income (Loss)
|
|||||
Consolidated Statements of Cash Flows |
|||||
Notes to Consolidated Financial Statements |
|||||
86
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of OPKO Health, Inc. and subsidiaries
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of OPKO Health, Inc. and subsidiaries (the Company) as of December 31, 2021 and 2020, the related consolidated statements of operations, comprehensive income (loss), shareholders' equity and cash flows for each of the three years in the period ended December 31, 2021, and the related notes and financial statement schedule included at Item 15(a)(1) (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2021, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated March 1, 2022 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
87
| Valuation of Goodwill and IPR&D for Rayaldee and Biologics | ||||||||
| Description of the Matter |
At December 31, 2021, the Company’s goodwill was $520.6 million, and indefinite lived in-process research and development assets (IPR&D) was $590.2 million. Included in the Rayaldee reporting unit was $86.6 million of goodwill. Included in the Biologics reporting unit was $139.8 million and $590.2 million of goodwill and IPR&D, respectively. As discussed in Note 3 to the consolidated financial statements, goodwill and indefinite lived IPR&D are tested at least annually for impairment or when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. To determine the estimated fair value of their reporting units and their intangible assets included within them, management considers both market and income valuation approaches.
Auditing management’s annual impairment tests for the goodwill and intangible assets in these reporting units was complex and highly judgmental due to the significant assumptions used in the determination of guideline companies, market transactions and market multiples, as well as the expected timing and amount of market revenue share and the discount rate used to estimate future cash flows, which are affected by expectations about future development of IPR&D, market, or economic conditions.
|
|||||||
| How We Addressed the Matter in Our Audit |
We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls over the Company’s annual goodwill and intangible assets impairment review process, including controls over management’s review of the significant assumptions in the Rayaldee and Biologics analysis described above.
To test the estimated fair value of the reporting units and the intangible assets included within them, we performed audit procedures that included, among others, assessing methodologies and testing the significant assumptions discussed above and the underlying data used by the Company in its analyses. We compared the significant assumptions used by management to current market and economic trends and other relevant factors. We involved valuation specialists to assist with assessing the methodologies and evaluating certain significant assumptions, such as the determination of guideline companies, market transactions, market multiples and the discount rates. We assessed the historical accuracy of management’s estimates and performed sensitivity analyses on significant assumptions to evaluate the changes in the fair value that would result from changes in the assumptions.
|
|||||||
| Variable Consideration in Determining Revenue from Services | ||||||||
| Description of the Matter |
For the year ended December 31, 2021, the Company recorded revenue from services of $1,607.1 million. As discussed in Note 15 to the consolidated financial statements, revenue from services includes amounts due under third-party and government payer programs, net of estimates for explicit and implicit price concessions and other elements of variable consideration. The Company estimates variable consideration by evaluating, among other factors, recent collections experience as well as changes in reimbursement regulations, claims processing and coverage determinations.
Auditing revenue from services is complex and highly judgmental due to the estimation required to measure the variable consideration. In particular, management applies judgment in evaluating whether changes in reimbursement regulations, claims processing and coverage determinations affect the estimate of the revenue management expects to be entitled to collect. This resulted in significant auditor judgment in the performance of our procedures.
|
|||||||
| How We Addressed the Matter in Our Audit |
We obtained an understanding, evaluated the design, and tested the operating effectiveness of controls over the Company’s variable consideration estimation process, including controls over management’s review of collections experience and the evaluation of factors that would affect the amount of variable consideration described above.
To test the estimate of variable consideration, we performed audit procedures that included, among others, assessing the methodology used and testing the underlying data used by the Company in its analysis. We compared the collection rates used by management to historical collection trends and evaluated whether changes in the regulatory environment or the Company’s business model, customer base, mix of services and other factors would affect the estimate of variable consideration. We assessed the historical accuracy of management’s estimate and performed sensitivity analyses to evaluate the changes in variable consideration that would result from changes in the expected collection rates used and the corresponding effect on revenue from services.
|
|||||||
88
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2007.
March 1, 2022
89
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of OPKO Health, Inc. and subsidiaries
Opinion on Internal Control over Financial Reporting
We have audited OPKO Health, Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2021, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, OPKO Health, Inc, and subsidiaries (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2021, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the 2021 consolidated financial statements of the Company and our report dated March 1, 2022 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Miami, Florida
March 1, 2022
90
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
| December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
| Accounts receivable, net | |||||||||||
| Inventory, net | |||||||||||
| Other current assets and prepaid expenses | |||||||||||
| Assets held for sale | |||||||||||
| Total current assets | |||||||||||
| Property, plant and equipment, net | |||||||||||
| Intangible assets, net | |||||||||||
| In-process research and development | |||||||||||
| Goodwill | |||||||||||
| Investments | |||||||||||
| Operating lease right-of-use assets | |||||||||||
| Other assets | |||||||||||
| Total assets | $ | $ | |||||||||
| LIABILITIES AND EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | $ | |||||||||
| Accrued expenses | |||||||||||
| Current maturities of operating leases | |||||||||||
| Liabilities associated with assets held for sale | |||||||||||
| Current portion of lines of credit and notes payable | |||||||||||
| Total current liabilities | |||||||||||
| Operating lease liabilities | |||||||||||
| Convertible notes | |||||||||||
| Deferred tax liabilities | |||||||||||
| Other long-term liabilities, principally contract liabilities, contingent consideration and lines of credit | |||||||||||
| Total long-term liabilities | |||||||||||
| Total liabilities | |||||||||||
| Equity: | |||||||||||
Common Stock - $ |
|||||||||||
Treasury Stock, - |
( |
( |
|||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive loss | ( |
( |
|||||||||
| Accumulated deficit | ( |
( |
|||||||||
| Total shareholders’ equity | |||||||||||
| Total liabilities and equity | $ | $ | |||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these statements.
91
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands, except share and per share data)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Revenues: | |||||||||||||||||
| Revenue from services | $ | $ | $ | ||||||||||||||
| Revenue from products | |||||||||||||||||
| Revenue from transfer of intellectual property and other | |||||||||||||||||
| Total revenues | |||||||||||||||||
| Costs and expenses: | |||||||||||||||||
| Cost of service revenue | |||||||||||||||||
| Cost of product revenue | |||||||||||||||||
| Selling, general and administrative | |||||||||||||||||
| Research and development | |||||||||||||||||
| Contingent consideration | ( |
( |
( |
||||||||||||||
| Amortization of intangible assets | |||||||||||||||||
| Asset impairment charges | |||||||||||||||||
| Gain on sale of assets | ( |
||||||||||||||||
| Total costs and expenses | |||||||||||||||||
| Operating income (loss) | ( |
||||||||||||||||
| Other income and (expense), net: | |||||||||||||||||
| Interest income | |||||||||||||||||
| Interest expense | ( |
( |
( |
||||||||||||||
| Fair value changes of derivative instruments, net | |||||||||||||||||
| Other income (expense), net | ( |
( |
|||||||||||||||
| Other income and (expense), net | ( |
( |
( |
||||||||||||||
| Income (loss) before income taxes and investment losses | ( |
( |
|||||||||||||||
| Income tax provision | ( |
( |
( |
||||||||||||||
| Net income (loss) before investment losses | ( |
( |
|||||||||||||||
| Loss from investments in investees | ( |
( |
( |
||||||||||||||
| Net income (loss) | $ | ( |
$ | $ | ( |
||||||||||||
| Income (loss) per share basic and diluted: | |||||||||||||||||
| Income (loss) per share | $ | ( |
$ | $ | ( |
||||||||||||
| Weighted average number of common shares outstanding, basic and diluted |
|||||||||||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these statements.
92
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(In thousands)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Net income (loss) | $ | ( |
$ | $ | ( |
||||||||||||
| Other comprehensive income (loss), net of tax: | |||||||||||||||||
| Change in foreign currency translation and other comprehensive income (loss) | ( |
( |
|||||||||||||||
| Comprehensive income (loss) | $ | ( |
$ | $ | ( |
||||||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these statements.
93
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF EQUITY
(In thousands, except share and per share data)
For the years ended December 31, 2021, 2020, 2019
| Common Stock | Treasury | Additional Paid-In Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total | ||||||||||||||||||||||||||||||||||||||||||
| Shares | Dollars | Shares | Dollars | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2018 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
| Equity-based compensation expense | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Exercise of common stock options and warrants | — | — | — | ( |
— | — | ( |
||||||||||||||||||||||||||||||||||||||||
| Adoption of ASU 2018-07 | — | — | — | — | ( |
— | |||||||||||||||||||||||||||||||||||||||||
| 2025 convertible notes including share lending agreement | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||
| Sale of common stock | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | ( |
( |
|||||||||||||||||||||||||||||||||||||||
| Other comprehensive loss | — | — | — | — | — | ( |
— | ( |
|||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2019 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
| Common Stock | Treasury | Additional Paid-In Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total | ||||||||||||||||||||||||||||||||||||||||||
| Shares | Dollars | Shares | Dollars | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2019 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
| Equity-based compensation expense | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Exercise of common stock options and warrants | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||
| — | — | — | — | — | — | ( |
( |
||||||||||||||||||||||||||||||||||||||||
| Net income | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2020 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
| Common Stock | Treasury | Additional Paid-In Capital |
Accumulated Other Comprehensive Loss |
Accumulated Deficit |
Total | ||||||||||||||||||||||||||||||||||||||||||
| Shares | Dollars | Shares | Dollars | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2020 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
| Equity-based compensation expense | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Exercise of common stock options and warrants | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||
| Conversion of 2025 convertible notes | ( |
— | — | — | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | ( |
( |
|||||||||||||||||||||||||||||||||||||||
| Other comprehensive loss | — | — | — | — | — | ( |
— | ( |
|||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2021 | $ | ( |
$ | ( |
$ | $ | ( |
$ | ( |
$ | |||||||||||||||||||||||||||||||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these statements.
94
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Cash flows from operating activities: | |||||||||||||||||
| Net income (loss) | $ | ( |
$ | $ | ( |
||||||||||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | |||||||||||||||||
| Depreciation and amortization | |||||||||||||||||
| Non-cash interest | |||||||||||||||||
| Amortization of deferred financing costs | |||||||||||||||||
| Losses from investments in investees | |||||||||||||||||
| Equity-based compensation – employees and non-employees | |||||||||||||||||
| Asset impairment charges | |||||||||||||||||
| Non-cash revenue from the transfer of intellectual property | ( |
||||||||||||||||
| Realized loss (gain) on disposal of fixed assets and sales of equity securities | ( |
( |
|||||||||||||||
| Loss on conversion of the 2025 Notes | |||||||||||||||||
| Change in fair value of equity securities and derivative instruments | ( |
||||||||||||||||
| Change in fair value of contingent consideration | ( |
( |
( |
||||||||||||||
| Deferred income tax provision | |||||||||||||||||
| Changes in assets and liabilities, net of the effects of acquisitions: | |||||||||||||||||
| Accounts receivable, net | ( |
||||||||||||||||
| Inventory, net | ( |
( |
|||||||||||||||
| Other current assets and prepaid expenses | ( |
||||||||||||||||
| Other assets | ( |
||||||||||||||||
| Accounts payable | ( |
||||||||||||||||
| Foreign currency measurement | ( |
( |
|||||||||||||||
| Contract liabilities | ( |
( |
( |
||||||||||||||
| Accrued expenses and other liabilities | ( |
||||||||||||||||
| Net cash provided by (used in) operating activities | ( |
||||||||||||||||
| Cash flows from investing activities: | |||||||||||||||||
| Investments in investees | ( |
( |
|||||||||||||||
| Proceeds from sale of investments | |||||||||||||||||
| Acquisition of businesses, net of cash acquired | ( |
||||||||||||||||
| Proceeds from the sale of property, plant and equipment | |||||||||||||||||
| Capital expenditures | ( |
( |
( |
||||||||||||||
| Net cash provided by (used in) investing activities | ( |
( |
|||||||||||||||
| Cash flows from financing activities: | |||||||||||||||||
| Issuance of common stock | |||||||||||||||||
| Issuance of 2023 Convertible Notes, including to related parties | |||||||||||||||||
| Debt issuance costs | ( |
( |
|||||||||||||||
| Proceeds from the exercise of common stock options and warrants | ( |
||||||||||||||||
| Borrowings on lines of credit | |||||||||||||||||
| Repayments of lines of credit | ( |
( |
( |
||||||||||||||
| Redemption of 2033 Senior Notes | ( |
||||||||||||||||
| Net cash (used in) provided by financing activities | ( |
( |
|||||||||||||||
| Effect of exchange rate changes on cash and cash equivalents | ( |
( |
|||||||||||||||
| Net increase (decrease) in cash and cash equivalents | ( |
( |
|||||||||||||||
| Cash and cash equivalents at beginning of period | |||||||||||||||||
| Cash and cash equivalents at end of period | $ | $ | $ | ||||||||||||||
| SUPPLEMENTAL INFORMATION: | |||||||||||||||||
| Interest paid | $ | $ | $ | ||||||||||||||
| Income taxes paid, net of refunds | $ | $ | ( |
$ | |||||||||||||
| Operating lease right-of-use assets due to adoption of ASU No. 2016-02 | $ | $ | $ | ||||||||||||||
| Operating lease liabilities due to adoption of ASU No. 2016-02 | $ | $ | $ | ||||||||||||||
| Operating lease right-of-use assets obtained in exchange for lease obligations | $ | $ | $ | ||||||||||||||
| Non-cash financing: | |||||||||||||||||
| Shares issued upon the conversion of: | |||||||||||||||||
| Common Stock options and warrants, surrendered in net exercise | $ | $ | $ | ||||||||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these statements.
95
OPKO Health, Inc. and Subsidiaries
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 Business and Organization
We are a diversified healthcare company that seeks to establish industry-leading positions in large and rapidly growing medical markets. Our diagnostics business includes BioReference Laboratories, Inc. (“BioReference”), one of the nation’s largest full service laboratories with an almost 250 -person sales and marketing team to drive growth and leverage new products. Our pharmaceutical business features Rayaldee, a U.S. Food and Drug Administration (“FDA”) approved treatment for secondary hyperparathyroidism (“SHPT”) in adults with stage 3 or 4 chronic kidney disease (“CKD”) and vitamin D insufficiency and a pipeline of products in various stages of development. Our leading product in development is Somatrogon (hGH-CTP), a once-weekly human growth hormone for which we have partnered with Pfizer, Inc. (“Pfizer”) and successfully completed a phase 3 study in August 2019. Regulatory applications for Somatrogon have been submitted to several countries around the world for review. In February 2022, the European Commission granted marketing authorization in the European Union for Somatrogon under the brand name NGENLA® to treat children and adolescents from as young as 3 years of age with growth disturbance due to insufficient secretion of growth hormone. In January 2022, the Ministry of Health, Labour and Welfare in Japan approved NGENLA® (Somatrogon) for the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone. In October 2021, Health Canada approved NGENLA® for the long-term treatment of pediatric patients who have growth hormone deficiency, and Australia’s Therapeutic Goods Administration approved NGENLA® for the long-term treatment of pediatric patients with growth disturbance due to insufficient secretion of growth hormone. We also submitted the initial Biologics License Application (“BLA”) with the FDA for approval of Somatrogon (hGH-CTP) in the United States and Pfizer received a Complete Response Letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States. We are incorporated in Delaware, and our principal executive offices are located in leased offices in Miami, Florida.
Through BioReference, we provide laboratory testing services, primarily to customers in the larger metropolitan areas in New York, New Jersey, Florida, Texas, Maryland, California, Pennsylvania, Delaware, Washington, DC, Illinois and Massachusetts, as well as to customers in a number of other states. We offer a comprehensive test menu of clinical diagnostics for blood, urine and tissue analysis. This includes hematology, clinical chemistry, immunoassay, infectious diseases, serology, hormones, and toxicology assays, as well as Pap smear, anatomic pathology (biopsies) and other types of tissue analysis. We market our laboratory testing services directly to physicians, geneticists, hospitals, clinics, correctional and other health facilities.
We operate established pharmaceutical platforms in Ireland, Chile, Spain, and Mexico, which are generating revenue and from which we expect to generate positive cash flow and facilitate future market entry for our products currently in development. In addition, we have a development and commercial supply pharmaceutical company and a global supply chain operation and holding company in Ireland. We own a specialty active pharmaceutical ingredients (“APIs”) manufacturer in Israel, which we expect will facilitate the development of our pipeline of molecules and compounds for our proprietary molecular diagnostic and therapeutic products.
Our research and development activities are primarily performed at facilities in Woburn, MA, Waterford, Ireland, Kiryat Gat, Israel, and Barcelona, Spain.
On January 18, 2022, Sema4 Holdings Corp. (“Sema4”) and OPKO announced they had signed an Agreement and Plan of Merger and Reorganization (the “GeneDx Merger Agreement”) with Sema4 Holdings Corp., a Delaware corporation (“Sema4”), pursuant to which Sema4 has agreed to acquire OPKO’s wholly owned subsidiary, GeneDx, Inc. (“GeneDx”), subject to satisfaction of customary closing conditions (the “GeneDx Transaction”). The GeneDx Transaction is expected to close in the second quarter of 2022.
Under the terms of the GeneDx Merger Agreement, Sema4 has agreed to acquire GeneDx for an upfront payment of $150 million in cash plus 80.0 million shares in Sema4, with up to an additional $150 million revenue-based milestones over the next two years (which will be payable in cash or Sema4 shares at Sema4’s discretion). Based on the closing stock price of Sema4 as of January 14, 2022, the total upfront consideration represents approximately $473 million, and the total aggregate consideration including potential milestones is approximately $623 million.
As of December 31, 2021, GeneDx met the held-for-sale accounting criteria and the related assets and liabilities are classified as held for sale in the consolidated balance sheet. Depending upon the value Sema4 shares upon closing of the transaction, an impairment charge may be incurred. GeneDx was included in our diagnostics segment as of December 31, 2021.
96
Note 2 Impact of COVID-19
As the disease caused by SARS-CoV-2, a novel strain of coronavirus, COVID-19 continues to spread and severely impact the U.S. economy and economies of other countries around the world, we continue to be a part of the coordinated public and private sector response to this unprecedented challenge as the COVID-19 pandemic continues. There continues to be a high level of uncertainty relating to how the pandemic will evolve, how governments and consumers will react, progress on the distribution of vaccines and whether the pandemic will have a longer-term effect on the healthcare industry and patient habits. In response to the COVID-19 pandemic, BioReference is providing COVID-19 solutions, including diagnostic molecular testing and serology antibody testing, to meet the testing needs of its numerous customer verticals, including physicians, health systems, long-term care facilities, governments, schools, employers, professional sports teams and entertainment venues, as well as the general public through relationships with retail pharmacy chains.
Revenue from services for the year ended December 31, 2021 increased by $344.9 million as compared to 2020 due to COVID-19 testing volumes. We are unable to predict how long the demand will continue for our COVID-19 related testing, or whether pricing and reimbursement policies for testing will sustain. In addition, in the second half of 2021, overall demand for COVID-19 testing has declined, and accordingly, the sustainability of our COVID-19 testing volumes is uncertain. Additionally, beginning in March 2020, BioReference experienced a decline in testing volumes due to the COVID-19 pandemic; however as stay at home orders and other restrictions have been lifted, we have seen our routine clinical and genomic testing volumes trending towards normalization with prior periods. Should stay at home orders or other restrictions be reenacted, we could see our routine testing levels decline. Excluding COVID-19 test volumes, for the year ended December 31, 2021, genomic and routine clinical test volume increased 26.4 % and 6.9 % as compared to volumes for the year ended December 31, 2020. Additionally, sales of Rayaldee have not increased in accordance with its expected growth trajectory as a result of challenges in onboarding new patients due to the COVID-19 pandemic. Federal, state and local governmental policies and initiatives designed to reduce the transmission of COVID-19 have resulted in, among other things, a significant reduction in physician office visits, the cancellation of elective medical procedures, customers closing or severely curtailing their operations (voluntarily or in response to government orders), and the adoption of work-from-home or shelter-in-place policies.
In March 2020, in response to the COVID-19 pandemic, the Coronavirus Aid, Relief, and Economic Security (CARES) Act was signed into law. The CARES Act provides numerous tax provisions and other stimulus measures, including temporary changes regarding the prior and future utilization of net operating losses, temporary changes to the prior and future limitations on interest deductions, temporary suspension of certain payment requirements for the employer portion of Social Security taxes, technical corrections from prior tax legislation for tax depreciation of certain qualified improvement property, and the creation of certain payroll tax credits associated with the retention of employees.
We have received, or expect to receive a number of benefits under the CARES Act including, but not limited to:
•During the year ended December 31, 2020, we received approximately $14 million under The Centers for Medicare & Medicaid Services (CMS) Accelerated and Advance Payment Program, which provides accelerated payments to Medicare providers/suppliers working to provide treatment to patients and combat the COVID-19 pandemic, and the amounts advanced are loans which will be offset against future claims and were repaid in 2021. These loans are initially recorded as contract liabilities included in Accrued expenses and are reduced as the amounts are recouped by CMS;
•We are eligible to defer depositing the employer’s share of Social Security taxes for payments due from March 27, 2020 through December 31, 2020, interest-free and penalty-free;
•We received approximately $16.2 million during 2020 from the funds that were distributed to healthcare providers for related expenses or lost revenues that are attributable to the COVID-19 pandemic. We recognized the $16.2 million grant in other revenues for the year ended December 31, 2020;
•U.S. Department of Health and Human Services (HHS), will provide claims reimbursement to healthcare providers generally at Medicare rates for testing uninsured patients; and
97
•Clinical laboratories are provided a one-year reprieve from the reporting requirements under the Protecting Access to Medicare Act (“PAMA”) as well as a one-year delay of reimbursement rate reductions for clinical laboratory services provided under Medicare that were scheduled to take place in 2021.
Since the pandemic began in the U.S., we have invested in testing capabilities and infrastructure to meet demand for our molecular and antibody testing for COVID-19. In 2021, we kicked off company-wide lab operations specimen acquisition, logistics, procurement, customer service, cost reduction initiatives to rightsize our cost structure to match the declining COVID testing volumes and to drive efficiency gains in our core clinical lines of business.
Three vaccines for COVID-19 have received approval or emergency authorization and have had increasingly widespread acceptance. However, we believe that, based on our experience with the pandemic, the high medical need for efficient and widespread testing for COVID-19 will extend beyond the current phase of the pandemic. Our belief is supported by the unprecedented healthcare and economic impact of the pandemic thus far, the uneven and incomplete rollout of vaccines and the fact that significant portions of the U.S. population may never be vaccinated, and the continued likelihood of surges of COVID-19 including from new strains of SARS-CoV-2 with uncertain susceptibility to the current vaccines. We believe that these factors have greatly magnified the need for more effective therapeutics, and the need for efficient and widespread testing, with properties targeted to the disease processes caused by serious viral infections.
Note 3 Summary of Significant Accounting Policies
Goodwill and intangible assets. Goodwill represents the difference between the purchase price and the estimated fair value of the net assets acquired accounted for by the acquisition method of accounting. Refer to Note 6. Goodwill, in-process research and development (“IPR&D”) and other intangible assets acquired in business combinations, licensing and other transactions at December 31, 2021 and 2020, was $1.4 billion and $1.7 billion, respectively.
Assets acquired and liabilities assumed in business combinations, licensing and other transactions are generally recognized at the date of acquisition at their respective fair values. Any excess of the purchase price over the estimated fair values of the net assets acquired is recognized as goodwill. At acquisition, we generally determine the fair value of intangible assets, including IPR&D, using the “income method.”
Subsequent to acquisition, goodwill and indefinite lived intangible assets are tested at least annually as of October 1 for impairment, or when events or changes in circumstances indicate it is more likely than not that the carrying amount of such assets may not be recoverable.
98
Goodwill was $520.6 million and $680.6 million, respectively, at December 31, 2021 and 2020. In addition, at December 31, 2021, Assets held for sale includes $151.8 million of goodwill related to GeneDx. Estimating the fair value of a reporting unit for goodwill impairment is highly sensitive to changes in projections and assumptions and changes in assumptions could potentially lead to impairment. We perform sensitivity analyses around our assumptions in order to assess the reasonableness of the assumptions and the results of our testing. Ultimately, potential changes in these assumptions may impact the estimated fair value of a reporting unit and result in an impairment if the fair value of such reporting unit is less than its carrying value.
Net intangible assets at December 31, 2021 and 2020, other than goodwill were $911.9 million and $1.1 billion, respectively, including IPR&D of $590.2
Upon obtaining regulatory approval, IPR&D assets are then accounted for as a finite-lived intangible asset and amortized on a straight-line basis over its estimated useful life. If the project is abandoned, the IPR&D asset is charged to expense. Finite lived intangible assets are tested for impairment when events or changes in circumstances indicate it is more likely than not that the carrying amount of such assets may not be recoverable. The testing includes a comparison of the carrying amount of the asset to its estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated undiscounted future cash flows, then an impairment charge is recognized for the amount by which the carrying amount of the asset exceeds the fair value of the asset.
We believe that our estimates and assumptions in testing goodwill and other intangible assets, including IPR&D, for impairment are reasonable and otherwise consistent with assumptions that marketplace participants would use in their estimates of fair value. However, if future results are not consistent with our estimates and assumptions, including as a result of the COVID-19 global pandemic, then we may be exposed to additional impairment charges, which could be material. Our 2021 impairment test of the OPKO Biologics reporting unit, including IPR&D related to Somatrogon, indicated an excess of estimated fair value over the carrying amount of approximately 19 %. We submitted the initial BLA with the FDA for approval of Somatrogon (hGH-CTP) in the United States and Pfizer received a Complete Response Letter in January 2022. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon in the United States. If we are unable to successfully commercialize Somatrogon in the U.S., or changes in projections and assumptions negatively impact our forecast of net cash flows, we may be exposed to a material impairment charge related to the IPR&D for Somatrogon.
99
Income taxes. Income taxes are accounted for under the asset-and-liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and the respective tax bases and for operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. We periodically evaluate the realizability of our net deferred tax assets. Our tax accruals are analyzed periodically and adjustments are made as events occur to warrant such adjustment. Valuation allowances on certain U.S. deferred tax assets and non-U.S. deferred tax assets are established, because realization of these tax benefits through future taxable income does not meet the more-likely-than-not threshold.
We operate in various countries and tax jurisdictions globally. For the year ended December 31, 2021, the tax rate differed from the U.S. federal statutory rate of 21% primarily due to the valuation allowance against certain U.S. and non-U.S. deferred tax assets, the relative mix in earnings and losses in the U.S. versus foreign tax jurisdictions, and the impact of certain discrete tax events and operating results in tax jurisdictions that do not result in a tax benefit.
Included in Other long-term liabilities is an accrual of $3.2 million related to uncertain tax positions involving income recognition. We recognize that local tax law is inherently complex and the local taxing authorities may not agree with certain tax positions taken. In connection with an examination of a 2014 and 2015 tax return in a foreign jurisdiction, the taxing authority has issued an initial income tax assessment of approximately $66 million (including interest). We are protesting this
100
The portion of our accounts receivable due from individual patients comprises the largest portion of credit risk. At December 31, 2021 and 2020, receivables due from patients represent approximately 1.7 % and 0.7 %, respectively, of our consolidated Accounts receivable, net.
101
Pending accounting pronouncements.
In August 2020, the FASB issued ASU No. 2020-06, “Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity's Own Equity (Subtopic 815-40).” ASU 2020-06 will simplify the accounting for convertible instruments by reducing the number of accounting models for convertible debt instruments and convertible preferred stock. The ASU is effective for public entities for fiscal years beginning after December 15, 2021, with early adoption permitted. We are currently evaluating the impact of this new guidance on our Consolidated Financial Statements.
Note 4 Income (loss) Per Share
102
A total of 62,204,391 , 70,029,480 and 67,765,380 potential shares of Common Stock have been excluded from the calculation of diluted net income (loss) per share for the years ended December 31, 2021, 2020 and 2019, respectively, because their inclusion would be antidilutive. A full presentation of diluted earnings per share has not been provided because the required adjustments to the numerator and denominator resulted in diluted earnings per share equivalent to basic earnings per share.
During the year ended December 31, 2021, 445,437 Common Stock options to purchase shares of our Common Stock were exercised, resulting in the issuance of 445,437 shares of Common Stock. Of the 445,437 Common Stock options exercised, 0 shares of Common Stock were surrendered in lieu of a cash payment via the net exercise feature of the agreements.
During the year ended December 31, 2020, 206,875 Common Stock options to purchase shares of our Common Stock were exercised, resulting in the issuance of 206,875 shares of Common Stock. Of the 206,875 Common Stock options exercised, 0 shares of Common Stock were surrendered in lieu of a cash payment via the net exercise feature of the agreements.
103
Note 5 Investments
Investments
The following table reflects the accounting method, carrying value and underlying equity in net assets of our unconsolidated investments as of December 31, 2021 and 2020:
| (in thousands) | As of December 31, 2021 | As of December 31, 2020 | ||||||||||||||||||||||||
| Investment type | Investment Carrying Value | Underlying Equity in Net Assets | Investment Carrying Value | Underlying Equity in Net Assets | ||||||||||||||||||||||
| Equity method investments | $ | $ | $ | $ | ||||||||||||||||||||||
| Variable interest entity, equity method | ||||||||||||||||||||||||||
| Equity securities | ||||||||||||||||||||||||||
| Equity securities with no readily determinable fair value | ||||||||||||||||||||||||||
| Warrants and options | ||||||||||||||||||||||||||
| Total carrying value of investments | $ | $ | ||||||||||||||||||||||||
Equity method investments
Our equity method investments consist of investments in Pharmsynthez (ownership 9 %), Cocrystal Pharma, Inc. (“COCP”) (3 %), Non-Invasive Monitoring Systems, Inc. (“NIMS”) (1 %), Neovasc Inc. (“Neovasc”) (1 %), InCellDx, Inc. (“InCellDx”) (29 %), BioCardia, Inc. (“BioCardia”) (1 %), and Xenetic Biosciences, Inc. (“Xenetic”) (1 %). The aggregate amount of assets, liabilities, and net losses of our equity method investees as of and for the year ended December 31, 2021 were $223.6 million, $37.9 million, and $69.4 million, respectively. The aggregate total assets, liabilities, and net losses of our equity method investees as of and for the year ended December 31, 2020 was $90.9 million, $28.4 million, and $75.4 million, respectively. We have determined that we and/or our related parties can significantly influence control of our equity method investments through our board representation and/or voting power. Accordingly, we account for our investment in these entities under the equity method and record our proportionate share of their losses in Loss from investments in investees in our Consolidated Statement of Operations. The aggregate value of our equity method investments based on the quoted market price of their respective shares of common stock and the number of shares held by us as of December 31, 2021 and 2020 was $4.5 million and 7.5 million, respectively.
Investments in Equity securities
Our equity securities consist of investments in Phio Pharmaceuticals (“Phio”) (ownership 0.01 %), VBI Vaccines Inc. (“VBI”) (1 %), ChromaDex Corporation (“ChromaDex”) (0.1 %), Eloxx Pharmaceuticals, Inc. (“Eloxx”) (2 %), CAMP4 Therapeutics Corporation (“CAMP4”) (4.58 %), and HealthSnap, Inc. (6.46 %). We have determined that our ownership, along with that of our related parties, does not provide us with significant influence over the operations of these investments. Accordingly, we account for our investment in these entities as equity securities, and we record changes in the fair value of these investments in Other income (expense) each reporting period when they have readily determinable fair value. Equity securities without a readily determinable fair value are adjusted to fair value when there is an observable price change. Net gains and losses on our equity securities for the year ended December 31, 2021, 2020 and 2019 are as follows:
| For the year ended December 31 | |||||||||||||||||
| (in thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Equity Securities: | |||||||||||||||||
| Net gains and (losses) recognized during the period on equity securities | $ | ( |
$ | $ | ( |
||||||||||||
| Less: Net gains realized during the period on equity securities | ( |
( |
|||||||||||||||
| Unrealized net gains and losses recognized during the period on equity securities still held at the reporting date | $ | ( |
$ | $ | ( |
||||||||||||
Sales of investments
Gains (losses) included in earnings from sales of our investments are recorded in Other income (expense), net in our Consolidated Statement of Operations. The cost of securities sold is based on the specific identification method.
104
Warrants and options
In addition to our equity method investments and equity securities, we hold options to purchase 47 thousand additional shares of BioCardia, all of which were vested as of December 31, 2021 and 2020, and 33 thousand, 0.7 million to purchase additional shares of COCP and InCellDx, Inc., respectively. We recorded the changes in the fair value of the options and warrants in Fair value changes of derivative instruments, net in our Consolidated Statement of Operations. We also recorded the fair value of the options and warrants in Investments, net in our Consolidated Balance Sheet. See further discussion of the Company’s options and warrants in Note 19 and Note 20.
Investments in variable interest entities
We have determined that we hold variable interests in LeaderMed Health Group Limited (“LeaderMed”), Detect Genomix, LLC (“Detect Genomix”) and Zebra Biologics, Inc. (“Zebra”). We made this determination as a result of our assessment that they do not have sufficient resources to carry out their principal activities without additional financial support.
On September 14, 2021, we and LeaderMed Health Group Limited (“LeaderMed”), a pharmaceutical development company with operations based in Asia, announced the formation of a joint venture to develop, manufacture and commercialize two of OPKO’s clinical stage, long-acting drug products in Greater China and eight other Asian territories. Under the terms of the agreements, we have granted the joint venture exclusive rights to develop, manufacture and commercialize (a) OPK88003, an oxyntomodulin analog being developed for the treatment of obesity and diabetes, and (b) Factor VIIa-CTP, a novel long-acting coagulation factor being developed to treat hemophilia, in exchange for 4,703 shares 47 % ownership interest in the joint venture. In addition, we received an upfront payment of $1.0 million and will be reimbursed for clinical trial material and technical support we provide the joint venture.
In order to determine the primary beneficiary of the joint venture, we evaluated our investment and our related parties’ investment, as well as our investment combined with the related parties’ investment to identify if we had the power to direct the activities that most significantly impact the economic performance of the joint venture. Based on the capital structure, governing documents and overall business operations of the joint venture, we determined that, while a VIE, we do not have the power to direct the activities that most significantly impact the joint venture’s economic performance and do not have an obligation to fund expected losses. We did determine, that we can significantly influence control of the joint venture through our board representation and voting power. Therefore, we have the ability to exercise significant influence over the joint venture’s operations and account for our investment in the joint venture under the equity method.
In August 2020, GeneDx, Inc., a subsidiary of BioReference, announced that it had entered into an agreement with Pediatrix Medical Group (“Pediatrix”), a provider of maternal-fetal, and pediatric medical and surgical subspecialty physician services, to offer genomic sequencing to support clinical diagnosis in neonatal intensive care units staffed by Pediatrix’s affiliated neonatologists. The offering is planned to include whole exome and whole genome sequencing and genomic support services under the brand Detect Genomix.
Our initial capital investment in Detect Genomix was $245,000 for which we received a 49 % ownership interest in Detect Genomix. We are required to make additional capital contributions to Detect Genomix in accordance with our percentage interests if Detect Genomix is unable to generate positive cash flow from operations or is unable to obtain alternative financing. We have not made any other investments in or loans to Detect Genomix through December 31, 2021. In January 2022, the Detect Genomix agreement was terminated.
In order to determine the primary beneficiary of Detect Genomix, we evaluated our investment to identify if we had the power to direct the activities that most significantly impact the economic performance of Detect Genomix. Based on the capital structure, governing documents and overall business operations of Detect Genomix, we determined that, while a VIE, we do not have the power to direct the activities that most significantly impact Detect Genomix’s economic performance. We determined, however, that we can significantly influence control of Detect Genomix through our board representation and voting power. Therefore, we have the ability to exercise significant influence over Detect Genomix’s operations and account for our investment in Detect Genomix under the equity method. The joint venture was dissolved in January 2022.
We own 1,260,000 shares of Zebra Series A-2 Preferred Stock and 900,000 shares of Zebra restricted common stock (ownership 29
In order to determine the primary beneficiary of Zebra, we evaluated our investment and our related parties’ investment, as well as our investment combined with the related parties’ investment to identify if we had the power to direct the activities that most significantly impact the economic performance of Zebra. Based on the capital structure, governing documents and
105
106
Note 6 Composition of Certain Financial Statement Captions
| For the years ended December 31, | |||||||||||
| (In thousands) | 2021 | 2020 | |||||||||
| Accounts receivable, net | |||||||||||
| Accounts receivable | $ | $ | |||||||||
| Less: allowance for doubtful accounts | ( |
( |
|||||||||
| $ | $ | ||||||||||
| Inventories, net | |||||||||||
| Consumable supplies | $ | $ | |||||||||
| Finished products | |||||||||||
| Work in-process | |||||||||||
| Raw materials | |||||||||||
| Less: inventory reserve | ( |
( |
|||||||||
| $ | $ | ||||||||||
| Other current assets and prepaid expenses | |||||||||||
| Prepaid supplies | $ | $ | |||||||||
| Prepaid insurance | |||||||||||
| Taxes recoverable | |||||||||||
| Other receivables | |||||||||||
| Other | |||||||||||
| $ | $ | ||||||||||
| Property, plant and equipment, net: | |||||||||||
| Machinery, medical and other equipment | $ | $ | |||||||||
| Leasehold improvements | |||||||||||
| Furniture and fixtures | |||||||||||
| Automobiles and aircraft | |||||||||||
| Software | |||||||||||
| Building | |||||||||||
| Land | |||||||||||
| Construction in process | |||||||||||
| Less: accumulated depreciation | ( |
( |
|||||||||
| $ | $ | ||||||||||
| Intangible assets, net: | |||||||||||
| Customer relationships | $ | $ | |||||||||
| Technologies | |||||||||||
| Trade names | |||||||||||
| Covenants not to compete | |||||||||||
| Licenses | |||||||||||
| Product registrations | |||||||||||
| Other | |||||||||||
| Less: accumulated amortization | ( |
( |
|||||||||
| $ | $ | ||||||||||
107
| For the years ended December 31, | |||||||||||
| (In thousands) | 2021 | 2020 | |||||||||
| Accrued expenses: | |||||||||||
| Inventory received but not invoiced | $ | $ | |||||||||
| Employee benefits | |||||||||||
| Commitments and contingencies | |||||||||||
| Clinical trials | |||||||||||
| Finance leases short-term | |||||||||||
| Professional fees | |||||||||||
| Contingent consideration | |||||||||||
| Contract liabilities | |||||||||||
| Other | |||||||||||
| $ | $ | ||||||||||
| Other long-term liabilities: | |||||||||||
| Finance leases long-term | $ | $ | |||||||||
| Contingent consideration | |||||||||||
| Mortgages and other debts payable | |||||||||||
| Contract liabilities | |||||||||||
| Other | |||||||||||
| $ | $ | ||||||||||
Our intangible assets and goodwill relate principally to our completed acquisitions of OPKO Renal, OPKO Biologics, EirGen and BioReference. We amortize intangible assets with definite lives on a straight-line basis over their estimated useful lives. The estimated useful lives by asset class are as follows: technologies - 7 -17 years, customer relationships - 7 -20 years, product registrations - 7 -10 years, covenants not to compete - 5 years, trade names - 5 -10 years, other 9 -13 years. We do not anticipate capitalizing the cost of product registration renewals, rather we expect to expense these costs, as incurred. Our goodwill is not tax deductible for income tax purposes in any jurisdiction in which we operate.
As of December 31, 2021, GeneDx met the held-for-sale accounting criteria and the related assets and liabilities are recognized at the lower of carrying value or fair value less costs to sell in the consolidated balance sheet. In addition, at December 31, 2021, Assets held for sale includes $151.8 million of goodwill related to GeneDx. The changes in value of the intangible assets and goodwill during the year ended December 31, 2020, are primarily due to foreign currency fluctuations between the Chilean Peso, the Euro and the Shekel against the U.S. dollar. The changes in value of the intangible assets and goodwill during the year ended December 31, 2019 are primarily due to an impairment charge of $44.8 million to write our IPR&D assets for OPK88003 and CURNA’s platform technology for oligonucleotide therapeutics down to their estimated fair value, a goodwill impairment charge of $26.2 million to write the carrying amount of the OPKO Diagnostics, CURNA and Transition Therapeutics reporting units down to their estimated fair value, and an impairment charge of $20.7 million to write our intangible asset for the Claros Analyzer down to its estimated fair value.
The following table reflects the changes in the allowance for doubtful accounts, provision for inventory reserve and tax valuation allowance accounts:
| (In thousands) | Beginning balance |
Charged to expense |
Written-off | Charged to other |
Ending balance |
||||||||||||||||||||||||
| 2021 | |||||||||||||||||||||||||||||
| Allowance for doubtful accounts | $ | ( |
( |
$ | ( |
||||||||||||||||||||||||
| Inventory reserve | $ | ( |
( |
$ | ( |
||||||||||||||||||||||||
| Tax valuation allowance | $ | ( |
$ | ( |
|||||||||||||||||||||||||
| 2020 | |||||||||||||||||||||||||||||
| Allowance for doubtful accounts | $ | ( |
( |
$ | ( |
||||||||||||||||||||||||
| Inventory reserve | $ | ( |
( |
$ | ( |
||||||||||||||||||||||||
| Tax valuation allowance | $ | ( |
( |
$ | ( |
||||||||||||||||||||||||
108
The following table summarizes the changes in Goodwill by reporting unit during the years ended December 31, 2021 and 2020.
| 2021 | 2020 | ||||||||||||||||||||||||||||||||||
| (In thousands) | Gross goodwill at January 1 | Cumulative impairment at January 1 | Goodwill impairment | Foreign exchange and other | Balance at December 31st | Gross goodwill at January 1 | Cumulative impairment at January 1 | Goodwill impairment | Foreign exchange and other | Balance at December 31st | |||||||||||||||||||||||||
| Pharmaceuticals | |||||||||||||||||||||||||||||||||||
| CURNA | $ | $ | ( |
$ | $ | $ | $ | $ | ( |
$ | $ | $ | |||||||||||||||||||||||
| Rayaldee | ( |
||||||||||||||||||||||||||||||||||
| FineTech | ( |
( |
|||||||||||||||||||||||||||||||||
| OPKO Biologics | |||||||||||||||||||||||||||||||||||
| OPKO Chile | ( |
||||||||||||||||||||||||||||||||||
| OPKO Health Europe | ( |
||||||||||||||||||||||||||||||||||
| OPKO Mexico | ( |
( |
|||||||||||||||||||||||||||||||||
| Transition Therapeutics | ( |
( |
|||||||||||||||||||||||||||||||||
| Diagnostics | |||||||||||||||||||||||||||||||||||
| BioReference | ( |
||||||||||||||||||||||||||||||||||
| OPKO Diagnostics | ( |
( |
|||||||||||||||||||||||||||||||||
| $ | $ | ( |
$ | $ | ( |
$ | $ | $ | ( |
$ | $ | $ | |||||||||||||||||||||||
Foreign exchange and other amounts for the year ended December 31, 2021 includes amounts related to GeneDx which is included as Assets held for sale at December 31, 2021.
109
Note 7 Debt
As of December 31, 2021 and 2020, our debt consists of the following:
| (In thousands) | As of December 31, 2021 | As of December 31, 2020 | |||||||||
| 2025 Notes | $ | $ | |||||||||
| 2023 Convertible Notes | |||||||||||
| 2033 Senior Notes | |||||||||||
| JP Morgan Chase | |||||||||||
| Chilean and Spanish lines of credit | |||||||||||
| Current portion of notes payable | |||||||||||
| Long term portion of notes payable | |||||||||||
| Total | $ | $ | |||||||||
| Balance sheet captions | |||||||||||
| Convertible Notes | $ | $ | |||||||||
| Current portion of lines of credit and notes payable | |||||||||||
| LT notes payable included in long-term liabilities | |||||||||||
| Total | $ | $ | |||||||||
On February 25, 2020, we entered into a credit agreement with an affiliate of Dr. Frost, pursuant to which the lender committed to provide us with an unsecured line of credit in the amount of $100 million. The line of credit called for a commitment fee equal to 0.25 % per annum of the unused portion of the line. No funds were borrowed under this line of credit and we terminated this line of credit in June 2021.
In February 2019, we issued $200.0 million aggregate principal amount of Senior Convertible Notes due 2025 (the “2025 Notes”) in an underwritten public offering. The 2025 Notes bear interest at a rate of 4.50 % per year, payable semiannually in arrears on February 15 and August 15 of each year. The 2025 Notes mature on February 15, 2025, unless earlier repurchased, redeemed or converted.
Holders may convert their 2025 Notes into shares of Common Stock at their option at any time prior to the close of business on the business day immediately preceding November 15, 2024 only under the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ended March 31, 2019 (and only during such calendar quarter), if the last reported sale price of our Common Stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130 % of the conversion price on each applicable trading day; (2) during the five business day period after any five consecutive trading day period (the “measurement period”) in which the trading price per $1,000 principal amount of 2025 Notes for each trading day of the measurement period was less than 98 % of the product of the last reported sale price of our Common Stock and the conversion rate on each such trading day; (3) if we call any or all of the 2025 Notes for redemption, at any time prior to the close of business on the scheduled trading day immediately preceding the redemption date; or (4) upon the occurrence of specified corporate events set forth in the indenture governing the 2025 Notes. On or after November 15, 2024, until the close of business on the business day immediately preceding the maturity date, holders of the 2025 Notes may convert their notes at any time, regardless of the foregoing conditions. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our Common Stock, or a combination of cash and shares of our Common Stock, at our election.
The initial and current conversion rate for the 2025 Notes is 236.7424 shares of Common Stock per $1,000 principal amount of 2025 Notes (equivalent to a conversion price of approximately $4.22 per share of Common Stock). The conversion rate for the 2025 Notes is subject to adjustment in certain events, but will not be adjusted for any accrued and unpaid interest. In addition, following certain corporate events that occur prior to the maturity date of the 2025 Notes or if we deliver a notice of redemption, in certain circumstances the indenture governing the 2025 Notes requires an increase in the conversion rate of the 2025 Notes for a holder who elects to convert its notes in connection with such a corporate event or notice of redemption, as the case may be.
We may not redeem the 2025 Notes prior to February 15, 2022. We may redeem for cash any or all of the notes, at our option, on or after February 15, 2022, if the last reported sale price of our Common Stock has been at least 130 % of the then current conversion price for the notes for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading day period (including the last trading day of such period) ending on, and including, the trading day immediately
110
preceding the date on which we provide notice of redemption at a redemption price equal to 100 % of the principal amount of the notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. No sinking fund is provided for the 2025 Notes.
If we undergo a fundamental change, as defined in the indenture governing the 2025 Notes, prior to the maturity date of the 2025 Notes, holders may require us to repurchase for cash all or any portion of their notes at a repurchase price equal to 100 % of the principal amount of the notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. The 2025 Notes are our senior unsecured obligations and rank senior in right of payment to any of our indebtedness that is expressly subordinated in right of payment to the 2025 Notes; equal in right of payment to any of our existing and future liabilities that are not so subordinated; effectively junior in right of payment to any of our secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all indebtedness and other liabilities (including trade payables) of our current or future subsidiaries.
In May 2021, we entered into exchange agreements with certain holders of the 2025 Notes pursuant to which the holders exchanged $55.4 million in aggregate principal amount of the outstanding 2025 Notes for 19,051,270 shares of our Common Stock (the “Exchange”). We recorded an $11.1 million non-cash loss related to the Exchange.
In conjunction with the issuance of the 2025 Notes, we agreed to loan up to 30,000,000 shares of our Common Stock to affiliates of the underwriter in order to assist investors in the 2025 Notes to hedge their position. Following consummation of the Exchange, the number of outstanding borrowed shares of Common Stock was reduced by 8,105,175 shares. As of December 31, 2021 and 2020, a total of 21,144,825 and 29,250,000 shares were issued under the share lending arrangement, respectively. We will not receive any of the proceeds from the sale of the borrowed shares, but we received a one-time nominal fee of $0.3 million for the newly issued shares. Shares of our Common Stock outstanding under the share lending arrangement are excluded from the calculation of basic and diluted earnings per share. See Note 4.
As required by ASC 470-20, “Debt with Conversion and Other Options,” we calculated the equity component of the 2025 Notes, taking into account both the fair value of the conversion option and the fair value of the share lending arrangement. The equity component was valued at $52.6 million at issue date and this amount was recorded as Additional paid-in capital, which resulted in a discount on the 2025 Notes. The discount is being amortized to Interest expense over the term of the 2025 Notes, which results in an effective interest rate on the 2025 Notes of 11.2 %.
The following table sets forth information related to the 2025 Notes which is included in our Consolidated Balance Sheet as of December 31, 2021:
| (In thousands) | 2025 Senior Notes | Discount | Debt Issuance Costs | Total | |||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | ( |
$ | ( |
$ | |||||||||||||||||
| Amortization of debt discount and debt issuance costs | — | ||||||||||||||||||||||
| Conversion | ( |
( |
|||||||||||||||||||||
| Balance at December 31, 2021 | $ | $ | ( |
$ | ( |
$ | |||||||||||||||||
In February 2018, we issued a series of 5 % Convertible Promissory Notes (the “2023 Convertible Notes”) in the aggregate principal amount of $55.0 million. The 2023 Convertible Notes mature 5 years from the date of issuance. Each holder of a 2023 Convertible Note has the option, from time to time, to convert all or any portion of the outstanding principal balance of such 2023 Convertible Note, together with accrued and unpaid interest thereon, into shares of our Common Stock at a conversion price of $5.00 per share of Common Stock. We may redeem all or any part of the then issued and outstanding 2023 Convertible Notes, together with accrued and unpaid interest thereon, pro rata among the holders, upon no fewer than 30 days, and no more than 60 days, notice to the holders. The 2023 Convertible Notes contain customary events of default and representations and warranties of OPKO.
Purchasers of the 2023 Convertible Notes included an affiliate of Dr. Phillip Frost, M.D., our Chairman and Chief Executive Officer, and Dr. Jane H. Hsiao, Ph.D., MBA, our Vice-Chairman and Chief Technical Officer.
In January 2013, we entered into note purchase agreements with respect to the issuance and sale of our 3.0 % Senior Notes due 2033 (the “2033 Senior Notes”) in a private placement exempt from registration under the Securities Act. We issued the 2033 Senior Notes on January 30, 2013. The 2033 Senior Notes, which totaled $175.0 million in original principal amount, bear interest at the rate of 3.0 % per year, payable semiannually on February 1 and August 1 of each year. The 2033 Senior Notes mature on February 1, 2033, unless earlier repurchased, redeemed or converted. Upon a fundamental change as defined in the indenture, governing the 2033 Senior Notes, subject to certain exceptions, the holders may require us to repurchase all or
111
any portion of their 2033 Senior Notes for cash at a repurchase price equal to 100 % of the principal amount of the 2033 Senior Notes being repurchased, plus any accrued and unpaid interest to but not including the related fundamental change repurchase date.
From 2013 to 2016, holders of the 2033 Senior Notes converted $143.2 million in aggregate principal amount into an aggregate of 21,539,873 shares of Common Stock. On February 1, 2019, approximately $28.8 million aggregate principal amount of 2033 Senior Notes were tendered by holders pursuant to such holders’ option to require us to repurchase the 2033 Senior Notes as set forth in the indenture, governing the 2033 Senior Notes, following which repurchase only $3.0 million aggregate principal amount of the 2033 Senior Notes remained outstanding. Holders of the remaining $3.0 million principal amount of the 2033 Senior Notes may require us to repurchase such notes for 100 % of their principal amount, plus accrued and unpaid interest, on February 1, 2023, on February 1, 2028, or following the occurrence of a fundamental change as described above.
The terms of the 2033 Senior Notes, include, among others: (i) rights to convert the notes into shares of our Common Stock, including upon a fundamental change; and (ii) a coupon make-whole payment in the event of a conversion by the holders of the 2033 Senior Notes on or after February 1, 2017 but prior to February 1, 2019. We determined that these specific terms were embedded derivatives. Embedded derivatives are required to be separated from the host contract, the 2033 Senior Notes, and carried at fair value when: (a) the embedded derivative possesses economic characteristics that are not clearly and closely related to the economic characteristics of the host contract; and (b) a separate, stand-alone instrument with the same terms would qualify as a derivative instrument. We concluded that the embedded derivatives within the 2033 Senior Notes met these criteria and, as such, were valued separate and apart from the 2033 Senior Notes and recorded at fair value each reporting period.
For accounting and financial reporting purposes, we combined these embedded derivatives and valued them together as one unit of accounting. In 2017, certain terms of the embedded derivatives expired pursuant to the original agreement and the embedded derivatives no longer met the criteria to be separated from the host contract and, as a result, the embedded derivatives were no longer required to be valued separate and apart from the 2033 Senior Notes and were reclassified to additional paid in capital.
In November 2015, BioReference and certain of its subsidiaries entered into a credit agreement with JPMorgan Chase Bank, N.A. (“CB”), as lender and administrative agent, as amended (the “Credit Agreement”). The Credit Agreement provides for a $75.0 million secured revolving credit facility and includes a $20.0 million sub-facility for swingline loans and a $20.0 million sub-facility for the issuance of letters of credit.
On August 30, 2021, the Credit Agreement was amended and restated (the “A&R Credit Agreement”). The A&R Credit Agreement is guaranteed by all of BioReference’s domestic subsidiaries. The A&R Credit Agreement is also secured by substantially all assets of BioReference and its domestic subsidiaries, as well as a non-recourse pledge by us of our equity interest in BioReference. Availability under the A&R Credit Agreement is based on a borrowing base composed of eligible accounts receivables of BioReference and certain of its subsidiaries, as specified therein. As of December 31, 2021, $64.8 million remained available for borrowing under the Credit Agreement. Principal under the Credit Agreement is due upon maturity on August 30, 2024.
At BioReference’s option, borrowings under the A&R Credit Agreement (other than swingline loans) bear interest at (i) the CB floating rate (defined as the higher of (a) the prime rate and (b) the LIBOR rate (adjusted for statutory reserve requirements for Eurocurrency liabilities) for an interest period of one month plus 2.50 %) plus an applicable margin of 0.75 % or (ii) the LIBOR rate (adjusted for statutory reserve requirements for Eurocurrency liabilities) plus an applicable margin of 1.75 %. Swingline loans will bear interest at the CB floating rate plus the applicable margin. The A&R Credit Agreement also calls for other customary fees and charges, including an unused commitment fee of 0.375 % if the average quarterly availability is 50% or more of the revolving commitment, or 0.25 % if the average quarterly availability is less than or equal to 50% of the revolving commitments.
As of December 31, 2021 and 2020, no amount and $7.1 million, respectively, was outstanding under the A&R Credit Agreement.
The A&R Credit Agreement contains customary covenants and restrictions, including, without limitation, covenants that require BioReference and its subsidiaries to maintain a minimum fixed charge coverage ratio if availability under the new credit facility falls below a specified amount and to comply with laws and restrictions on the ability of BioReference and its subsidiaries to incur additional indebtedness or to pay dividends and make certain other distributions to the Company, subject to certain exceptions as specified therein. Failure to comply with these covenants would constitute an event of default under the A&R Credit Agreement, notwithstanding the ability of BioReference to meet its debt service obligations. The A&R Credit Agreement also includes various customary remedies for the lenders following an event of default, including the acceleration of
112
repayment of outstanding amounts under the A&R Credit Agreement and execution upon the collateral securing obligations under the A&R Credit Agreement. Substantially all the assets of BioReference and its subsidiaries are restricted from sale, transfer, lease, disposal or distributions to the Company, subject to certain exceptions. As of December 31, 2021, BioReference and its subsidiaries had net assets of approximately $1,103.6 million, which included goodwill of $283.0 million and intangible assets of $204.4 million.
In addition to the A&R Credit Agreement with CB, we had line of credit agreements with eleven other financial institutions as of December 31, 2021 and 2020 in the U.S., Chile and Spain. These lines of credit are used primarily as a source of working capital for inventory purchases.
The following table summarizes the amounts outstanding under the BioReference, Chilean and Spanish lines of credit:
| (Dollars in thousands) | Balance Outstanding | |||||||||||||||||||||||||
| Lender | Interest rate on borrowings at December 31, 2021 |
Credit line capacity |
December 31, 2021 |
December 31, 2020 |
||||||||||||||||||||||
| JP Morgan Chase | $ | $ | $ | |||||||||||||||||||||||
| Itau Bank | ||||||||||||||||||||||||||
| Bank of Chile | ||||||||||||||||||||||||||
| BICE Bank | ||||||||||||||||||||||||||
| Security Bank | ||||||||||||||||||||||||||
| Estado Bank | ||||||||||||||||||||||||||
| Santander Bank | ||||||||||||||||||||||||||
| Scotiabank | ||||||||||||||||||||||||||
| BCI Bank | ||||||||||||||||||||||||||
| Corpbanca | ||||||||||||||||||||||||||
| Banco De Sabadell | ||||||||||||||||||||||||||
| Banco Bilbao Vizcaya | ||||||||||||||||||||||||||
| Total | $ | $ | $ | |||||||||||||||||||||||
At December 31, 2021 and 2020, the weighted average interest rate on our lines of credit was approximately 5.4 % and 4.9 %, respectively.
At December 31, 2021 and 2020, we had notes payable and other debt (excluding the 2033 Senior Notes, the 2023 Convertible Notes, the 2025 Notes, the Credit Agreement and amounts outstanding under lines of credit described above) as follows:
| (In thousands) | December 31, 2021 |
December 31, 2020 |
|||||||||
| Current portion of notes payable | $ | $ | |||||||||
| Other long-term liabilities | |||||||||||
| Total | $ | $ | |||||||||
Note 8 Shareholders’ Equity
Our authorized capital stock consists of 1,000,000,000 shares of Common Stock, par value $0.01 per share, and 10,000,000 shares of Preferred Stock, par value $0.01 per share.
Sales of Common Stock
On October 29, 2019, we issued 50 million shares of our Common Stock at a price of $1.50 per share in an underwritten public offering (the “2019 Stock Offering”), resulting in net proceeds to the Company of approximately $70 million, after deducting underwriting commissions and offering expenses. In November 2019, pursuant to an option the Company granted the underwriters, we issued an additional 4,227,749 shares at $1.50 per share, less underwriting discounts and commissions,
113
resulting in net proceeds of approximately $6 million. Drs. Frost and Hsiao and Mr. Steven Rubin, members of OPKO’s senior management purchased an aggregate of 2,415,000 shares in the 2019 Stock Offering.
Common Stock
Subject to the rights of the holders of any shares of Preferred Stock currently outstanding or which may be issued in the future, the holders of the Common Stock are entitled to receive dividends from our funds legally available when, as and if declared by our Board of Directors, and are entitled to share ratably in all of our assets available for distribution to holders of Common Stock upon the liquidation, dissolution or winding-up of our affairs subject to the liquidation preference, if any, of any then outstanding shares of Preferred Stock. Holders of our Common Stock do not have any preemptive, subscription, redemption or conversion rights. Holders of our Common Stock are entitled to one vote per share on all matters which they are entitled to vote upon at meetings of stockholders or upon actions taken by written consent pursuant to Delaware corporate law. The holders of our Common Stock do not have cumulative voting rights, which means that the holders of a plurality of the outstanding shares can elect all of our directors. All of the shares of our Common Stock currently issued and outstanding are fully-paid and nonassessable. No dividends have been paid to holders of our Common Stock since our incorporation, and no cash dividends are anticipated to be declared or paid on our Common Stock in the reasonably foreseeable future.
Preferred Stock
Under our certificate of incorporation, our Board of Directors has the authority, without further action by stockholders, to designate up to 10 million shares of Preferred Stock in one or more series and to fix or alter, from time to time, the designations, powers and rights of each series of Preferred Stock and the qualifications, limitations or restrictions of any series of Preferred Stock, including dividend rights, dividend rate, conversion rights, voting rights, rights and terms of redemption (including sinking fund provisions), redemption price or prices, and the liquidation preference of any wholly issued series of Preferred Stock, any or all of which may be greater than the rights of the Common Stock, and to establish the number of shares constituting any such series.
Of the authorized Preferred Stock, 4,000,000 shares, 500,000 shares and 2,000,000 shares were designated Series A Preferred Stock, Series C Preferred Stock and Series D Preferred Stock, respectively. As of December 31, 2021 and 2020, there were no
Note 9 Accumulated Other Comprehensive Income (Loss)
For the year ended December 31, 2021, changes in Accumulated other comprehensive income (loss), net of tax, were as follows:
| (In thousands) | Foreign currency translation |
||||
| Balance at December 31, 2020 | $ | ( |
|||
| Other comprehensive loss | ( |
||||
| Balance at December 31, 2021 | $ | ( |
|||
For the year ended December 31, 2020, changes in Accumulated other comprehensive income, net of tax, were as follows:
| (In thousands) | Foreign currency translation |
||||
| Balance at December 31, 2019 | $ | ( |
|||
| Other comprehensive income | |||||
| Balance at December 31, 2020 | $ | ( |
|||
Note 10 Equity-Based Compensation
114
Modigene Plan are exercisable for a period of up to 10 years from date of grant. Vesting periods range from immediate to 5 years.
We classify the cash flows resulting from the tax benefit that arises when the tax deductions exceed the compensation cost recognized for those equity awards (excess tax benefits) as cash flows from operations. There were no
Valuation and Expense Information
We recorded equity-based compensation expense of $13.6 million, $8.9 million and $13.4 million for the years ended December 31, 2021, 2020, and 2019, respectively, all of which were reflected as operating expenses. Of the $13.6 million of equity based compensation expense recorded for the year ended December 31, 2021, $10.4 million was recorded as selling, general and administrative expenses, $1.9 million was recorded as research and development expenses and $1.3 million was recorded as a cost of revenue. Of the $8.9 million of equity based compensation expense recorded for the year ended December 31, 2020, $6.8 million was recorded as selling, general and administrative expense, $1.8 million was recorded as research and development expenses and $0.3 million was recorded as a cost of revenue. Of the $13.4 million of equity based compensation expense recorded for the year ended December 31, 2019, $9.7 million was recorded as selling, general and administrative expense, $2.0 million was recorded as research and development expenses and $1.6 million was recorded as cost of revenue.
As of December 31, 2021, there was $33.3 million of unrecognized compensation cost related to the stock options granted under our equity-based incentive compensation plans. Such cost is expected to be recognized over a weighted-average period of approximately 1.82 years.
Stock Options
We estimate the fair value of each stock option on the date of grant using the Black-Scholes-Merton Model option-pricing formula and amortize the fair value to expense over the stock option’s vesting period using the straight-line attribution approach. We account for forfeitures as they occur and apply the following assumptions in our Black-Scholes-Merton Model option-pricing formula:
| Year Ended December 31, 2021 |
Year Ended December 31, 2020 |
Year Ended December 31, 2019 |
|||||||||||||||
| Expected term (in years) | |||||||||||||||||
| Risk-free interest rate | |||||||||||||||||
| Expected volatility | |||||||||||||||||
| Expected dividend yield | |||||||||||||||||
Expected Term: For the expected term of options grants, we used an estimate of the expected option life based on historical experience.
Risk-Free Interest Rate: The risk-free interest rate is based on the rates paid on securities issued by the U.S. Treasury with a term approximating the expected life of the option.
Expected Volatility: The expected volatility for stock options was based on the historical volatility of our Common Stock.
Expected Dividend Yield: We do not intend to pay dividends on Common Stock for the foreseeable future. Accordingly, we used a dividend yield of zero in the assumptions.
We maintain incentive stock plans that provide for the grants of stock options to our directors, officers, employees and non-employee consultants. As of December 31, 2021, there were 11,555,335 shares of Common Stock reserved for issuance under our equity-based incentive plans. We intend to issue new shares upon the exercise of stock options. Stock options granted under these plans have been granted at an option price equal to the closing market value of the stock on the date of the grant. Stock options granted under these plans to employees typically become exercisable over four years in equal annual installments after the date of grant, and stock options granted to non-employee directors become exercisable in full one-year after the grant date, subject to, in each case, continuous service with us during the applicable vesting period. We assumed stock options to grant Common Stock as part of the mergers with Acuity Pharmaceuticals, Inc., Froptix, Inc., OPKO Biologics and BioReference, which reflected various vesting schedules, including monthly vesting to employees and non-employee consultants.
115
A summary of option activity under our stock option plans as of December 31, 2021, and the changes during the year is presented below:
| Options | Number of options |
Weighted average exercise price |
Weighted average remaining contractual term (years) |
Aggregate intrinsic value (in thousands) |
|||||||||||||||||||
| Outstanding at December 31, 2020 | $ | $ | |||||||||||||||||||||
| Granted | $ | ||||||||||||||||||||||
| Exercised | ( |
$ | |||||||||||||||||||||
| Forfeited | ( |
$ | |||||||||||||||||||||
| Expired | ( |
$ | |||||||||||||||||||||
| Outstanding at December 31, 2021 | $ | $ | |||||||||||||||||||||
| Vested and expected to vest at December 31, 2021 | $ | $ | |||||||||||||||||||||
| Exercisable at December 31, 2021 | $ | $ | |||||||||||||||||||||
The total intrinsic value of stock options exercised for the years ended December 31, 2021, 2020, and 2019 was $0.8 million, $0.4 million and $0.1 million, respectively.
The weighted average grant date fair value of stock options granted for the years ended December 31, 2021, 2020, and 2019 was $2.68 , $1.39 , and $1.15 , respectively. The total fair value of stock options vested during the years ended December 31, 2021, 2020, and 2019 was $8.1 million, $10.6 million and $18.6 million, respectively.
Note 11 Income Taxes
We operate and are required to file tax returns in the U.S. and various foreign jurisdictions.
The benefit (provision) for incomes taxes consists of the following:
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Current | |||||||||||||||||
| Federal | $ | $ | ( |
$ | |||||||||||||
| State | ( |
( |
|||||||||||||||
| Foreign | ( |
( |
( |
||||||||||||||
| ( |
( |
( |
|||||||||||||||
| Deferred | |||||||||||||||||
| Federal | ( |
( |
|||||||||||||||
| State | |||||||||||||||||
| Foreign | ( |
( |
( |
||||||||||||||
| ( |
( |
( |
|||||||||||||||
| Total, net | $ | ( |
$ | ( |
$ | ( |
|||||||||||
116
| (In thousands) | December 31, 2021 | December 31, 2020 | |||||||||
| Deferred income tax assets: | |||||||||||
| Federal net operating loss | $ | $ | |||||||||
| State net operating loss | |||||||||||
| Foreign net operating loss | |||||||||||
| Research and development expense | |||||||||||
| Tax credits | |||||||||||
| Stock options | |||||||||||
| Accruals | |||||||||||
| Equity investments | |||||||||||
| Bad debts | |||||||||||
| Lease liability | |||||||||||
| Foreign credits | |||||||||||
| Available-for-sale securities | |||||||||||
| Operating lease asset | |||||||||||
| Other | |||||||||||
| Deferred income tax assets | |||||||||||
| Deferred income tax liabilities: | |||||||||||
| Intangible assets | ( |
( |
|||||||||
| Convertible debt | ( |
( |
|||||||||
| Operating lease liability | ( |
( |
|||||||||
| Investment in subsidiaries | ( |
||||||||||
| Fixed assets | ( |
( |
|||||||||
| Other | ( |
( |
|||||||||
| Deferred income tax liabilities | ( |
( |
|||||||||
| Net deferred income tax assets (liabilities) | |||||||||||
| Valuation allowance | ( |
( |
|||||||||
| Net deferred income tax liabilities | $ | ( |
$ | ( |
|||||||
Note: Net deferred income tax liability balance includes $4.3 million recorded to Other Assets and $1.5 million recorded to Assets Held for Sale on the Consolidated Balance Sheet.
As of December 31, 2021, we have federal, state and foreign net operating loss carryforwards of approximately $470.2 million, $774.1 million and $86.8 million, respectively, that expire at various dates through 2041 unless indefinite in nature. As of December 31, 2021, we have research and development tax credit carryforwards of approximately $22.9 million that expire in varying amounts through 2041. As of each reporting date, management considers new evidence, both positive and negative, that could affect its view of the future realization of deferred tax assets. We have determined a valuation allowance is required against all of our net deferred tax assets that we do not expect to be utilized by the reversing of deferred income tax liabilities.
In 2020 we completed the transfer of certain assets to an OPKO affiliate. The transaction gave rise to a deferred tax asset of approximately $148.9 million. Realizability of a deferred tax asset ultimately depends on the existence of sufficient taxable income in the carryback and carryforward periods as permitted by tax law. The Company evaluated the realizability of the deferred tax asset as required by ASC 740-10-30-18. The Company has determined that the deferred tax asset is not more-likely-than-not to be realized as of December 31, 2020. As a result, the Company has recorded a full valuation allowance against the deferred tax asset.
Under Section 382 of the Internal Revenue Code of 1986, as amended, certain significant changes in ownership may restrict the future utilization of our income tax loss carryforwards and income tax credit carryforwards in the U.S. The annual limitation is equal to the value of our stock immediately before the ownership change, multiplied by the long-term tax-exempt rate (i.e., the highest of the adjusted federal long-term rates in effect for any month in the three-calendar-month period ending with the calendar month in which the change date occurs). This limitation may be increased under the IRC Section 338
117
Approach (IRS approved methodology for determining recognized Built-In Gain). As a result, federal net operating losses and tax credits may expire before we are able to fully utilize them.
During 2008, we conducted a study to determine the impact of the various ownership changes that occurred during 2007 and 2008. As a result, we have concluded that the annual utilization of our net operating loss carryforwards (“NOLs”) and tax credits is subject to a limitation pursuant to Internal Revenue Code Section 382. Under the tax law, such NOLs and tax credits are subject to expiration from 15 to 20 years after they were generated. As a result of the annual limitation that may be imposed on such tax attributes and the statutory expiration period, some of these tax attributes may expire prior to our being able to use them. There is no current impact on these financial statements as a result of the annual limitation. This study did not conclude whether OPKO’s predecessor, eXegenics, pre-merger NOLs were limited under Section 382. As such, of the $470.2 million of federal net operating loss carryforwards, at least approximately $41.0 million may not be able to be utilized.
During 2020, we conducted a study to determine whether any ownership changes occurred from 2009 through 2020. In 2021, the study has been updated and we have concluded that the annual utilization of our NOLs and tax credits is not subject to a limitation pursuant to Internal Revenue Code Section 382.
We file federal income tax returns in the U.S. and various foreign jurisdictions, as well as with various U.S. states and the Ontario and Nova Scotia provinces in Canada. We are subject to routine tax audits in all jurisdictions for which we file tax returns. Tax audits by their very nature are often complex and can require several years to complete. It is reasonably possible that some audits will close within the next twelve months, which we do not believe would result in a material change to our accrued uncertain tax positions.
U.S. Federal: Under the tax statute of limitations applicable to the Internal Revenue Code, we are no longer subject to U.S. federal income tax examinations by the Internal Revenue Service for years before 2018. However, because we are carrying forward income tax attributes, such as net operating losses and tax credits from those years, these attributes can still be audited when utilized on returns filed in the future.
State: Under the statute of limitations applicable to most state income tax laws, we are no longer subject to state income tax examinations by tax authorities for years before 2017 in states in which we have filed income tax returns. Certain states may take the position that we are subject to income tax in such states even though we have not filed income tax returns in such states and, depending on the varying state income tax statutes and administrative practices, the statute of limitations in such states may extend to years before 2017.
Foreign: Under the statute of limitations applicable to our foreign operations, we are generally no longer subject to tax examination for years before 2016 in jurisdictions where we have filed income tax returns.
Tax Cuts and Jobs Act
On December 22, 2017, the 2017 Tax Act was enacted into law and the new legislation contains several key tax provisions, including a reduction of the corporate income tax rate from 35% to 21% effective January 1, 2018 and a one-time mandatory transition tax on accumulated foreign earnings, among others. We were required to recognize the effect of the tax law changes in the period of enactment, such as remeasuring our U.S. deferred tax assets and liabilities, as well as reassessing the net realizability of our deferred tax assets and liabilities.
Effective January 1, 2018, the Tax Act provides for a new GILTI provision. Under the GILTI provision, certain foreign subsidiary earnings in excess of an allowable return on the foreign subsidiary’s tangible assets are included in U.S. taxable income. The Company’s GILTI inclusion is immaterial for the year ended December 31, 2021. The Company has not recorded any deferred taxes for future GILTI inclusions as any future inclusions are expected to be treated as a period expense and offset by net operating loss carryforwards in the U.S.
118
Unrecognized Tax Benefits
As of December 31, 2021, 2020, and 2019, the total amount of gross unrecognized tax benefits was approximately $11.5 million, $14.0 million, and $17.2 million, respectively. As of December 31, 2021, the total amount of unrecognized tax benefits that, if recognized, would affect our effective income tax rate was $(7.9 ) million. We account for any applicable interest and penalties on uncertain tax positions as a component of income tax expense and we recognized $0.2 million and $(0.1 ) million of interest expense for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2020 and 2019, $(10.0 ) million and $(13.2 ) million of the unrecognized tax benefits, if recognized, would have affected our effective income tax rate. We believe it is reasonably possible that up to $3.2 million of unrecognized tax benefits may be recognized within the next twelve months, mainly due to an expected audit settlement.
The following summarizes the changes in our gross unrecognized income tax benefits.
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Unrecognized tax benefits at beginning of period | $ | $ | $ | ||||||||||||||
| Gross increases – tax positions in current period | |||||||||||||||||
| Gross decreases – tax positions in prior period | ( |
( |
( |
||||||||||||||
| Gross decreases – settlements with taxing authorities | ( |
( |
|||||||||||||||
| Lapse of Statute of Limitations | ( |
( |
( |
||||||||||||||
| Unrecognized tax benefits at end of period | $ | $ | $ | ||||||||||||||
Other Income Tax Disclosures
The significant elements contributing to the difference between the federal statutory tax rate and the effective tax rate are as follows:
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Federal statutory rate | % | % | % | ||||||||||||||
| State income taxes, net of federal benefit | ( |
% | % | % | |||||||||||||
| Foreign income tax | % | % | ( |
% | |||||||||||||
| Income Tax Refunds | % | ( |
% | % | |||||||||||||
| Research and development tax credits | % | ( |
% | % | |||||||||||||
| Valuation allowance | % | % | ( |
% | |||||||||||||
| Rate change effect | ( |
% | % | % | |||||||||||||
| Non-deductible items | ( |
% | % | ( |
% | ||||||||||||
| Unrecognized tax benefits | % | ( |
% | % | |||||||||||||
| Impairments | % | % | ( |
% | |||||||||||||
| IPR&D benefit | % | ( |
% | % | |||||||||||||
| Stock options excess tax benefit | ( |
% | % | % | |||||||||||||
| Imputed interest | ( |
% | % | % | |||||||||||||
| Investment in subsidiaries | ( |
% | % | % | |||||||||||||
| Other | % | % | % | ||||||||||||||
| Total | ( |
% | % | ( |
% | ||||||||||||
119
Certain operations in Israel have been granted "Beneficiary Enterprise" status by the Israeli Income Tax Authority, which makes us eligible for tax benefits under the Israeli Law for Encouragement of Capital Investments, 1959. Under the terms of the Beneficiary Enterprise program, beneficiary income that is attributable to our operations in Kiryat Gat, Israel will be exempt from income tax through 2023. The impact of the tax holiday on a per share basis for the year ended December 31, 2021 was a benefit of $0.02 per share.
The following table reconciles our income (loss) before income taxes between U.S. and foreign jurisdictions:
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Pre-tax income (loss): | |||||||||||||||||
| U.S. | $ | ( |
$ | $ | ( |
||||||||||||
| Foreign | ( |
( |
( |
||||||||||||||
| Total | $ | ( |
$ | $ | ( |
||||||||||||
In 2021, we revised our position regarding unrepatriated foreign earnings to a partially reinvested assertion. We assert that all foreign earnings will be indefinitely reinvested, with the exception of certain foreign investments in which earnings and cash generation are in excess of local needs. With the passage of the Tax Act, dividends of earnings from non-U.S. operations are generally no longer subject to U.S. income tax. We continue to analyze and adjust the estimated impact of the non-U.S. income and withholding tax liabilities based on the source of these earnings, as well as the expected means through which those earnings may be taxed. We have accrued a withholding tax estimate of $1.8 million related to earnings that are not deemed to be permanently reinvested.
Note 12 Related Party Transactions
In August 2020, we paid a $125,000 filing fee to the Federal Trade Commission (the “FTC”) in connection with filings made by us and Dr. Jane Hsiao, our Vice Chairman and Chief Technical Officer, under the Hart-Scott-Rodino Antitrust Improvements Act of 1976 (“HSR Act”) relating to her percentage equity ownership interest in OPKO and potential future purchases of our Common Stock.
In August 2020, Dr. Phillip Frost, our Chairman and Chief Executive Officer, paid a filing fee of $280,000 to the FTC under the HSR Act in connection with filings made by us and Dr. Frost, relating to his percentage equity ownership interest in OPKO and potential future purchases of our Common Stock. We reimbursed Dr. Frost for the HSR filing fee.
In August 2020, GeneDx entered into an agreement with Mednax Services, Inc. (“Mednax Services”), a subsidiary of MEDNAX, Inc., (“MEDNAX”) pursuant to which the parties formed a joint venture under the brand Detect Genomix. GeneDx’s initial capital investment in Detect Genomix was $245,000 for which GeneDx received a 49 % ownership interest in Detect Genomix, and Mednax Services contributed $255,000 in exchange for a 51 % ownership interest in Detect Genomix. Adam Logal, the Company’s CFO, was the chair and sat on the Board of Managers of the joint venture. Mednax Services provided administrative services to the joint venture pursuant to an administrative services agreement. GeneDx provided laboratory services to the joint venture. Dr. Roger Medel, a director of the Company, is the former Chief Executive Officer of MEDNAX and Mednax Services. Dr. Medel continues to serve on the board of MEDNAX. The joint venture was dissolved in January 2022.
On February 25, 2020, we entered into a credit agreement with an affiliate of Dr. Frost, pursuant to which the lender committed to provide us with an unsecured line of credit in the amount of $100 million. This line of credit called for a commitment fee equal to 0.25 % per annum of the unused portion of the line. We terminated this line of credit in June 2021 and as of December 31, 2021, no amount was outstanding thereunder.
On October 29, 2019, we issued 50 million shares of our Common Stock at a price of $1.50 per share in the 2019 Stock Offering, resulting in net proceeds to the Company of approximately $70 million, after deducting underwriting commissions and offering expenses. In November 2019, pursuant to an option the Company granted the underwriters, we issued an additional 4,227,749 shares at the public offering price, less underwriting discounts and commissions, resulting in net proceeds to the Company of approximately $6 million. Drs. Frost and Hsiao and Mr. Steven Rubin, members of OPKO’s senior management purchased an aggregate of 2,415,000 shares of Common Stock in the 2019 Stock Offering.
On March 1, 2019, OPKO Pharmaceuticals, LLC entered into an assignment agreement with Xenetic Biosciences, Inc., as amended from time to time (the “Assignment Agreement”), pursuant to which Xenetic acquired all of OPKO Pharmaceuticals’ right, title and interest in and to that certain Intellectual Property License Agreement (the “IP License Agreement”), entered into between The Scripps Research Institute and OPKO Pharmaceuticals, regarding certain patents for novel CAR T platform
120
technology and through which the Scripps Research Institute granted an exclusive royalty-bearing license in exchange for royalties, subject to the terms of the IP License Agreement.
Under the Assignment Agreement and the IP License Agreement, Xenetic issued to OPKO Pharmaceuticals 164,062 shares of Xenetic common stock (the “OPKO Transaction Shares”). In connection with the Assignment Agreement, OPKO Pharmaceuticals entered into a voting agreement pursuant to which OPKO Pharmaceuticals agreed, among other things, to vote its shares in Xenetic in favor of the transactions contemplated by the Assignment Agreement, and a lock-up agreement with Xenetic which restricts OPKO Pharmaceuticals’ sale or transfer of any of the OPKO Transaction Shares as provided therein and as otherwise required by law. The Assignment Agreement and the obligations thereunder took effect on July 19, 2019, after Xenetic satisfied certain closing conditions, including obtaining stockholder approval and securing certain financing.
The Company owns approximately 9 % of Pharmsynthez, and Pharmsynthez is Xenetic’s largest and controlling stockholder. Dr. Richard Lerner, a director of the Company until his death on December 2, 2021, was a co-inventor of Xenetic’s technology and received 31,240 shares of Xenetic upon the closing of the Xenetic transactions described above. Adam Logal, our Senior Vice President and Chief Financial Officer, is a director of Xenetic.
In March 2019, we paid the $125,000 filing fee to the FTC in connection with filings made by us and Dr. Jane Hsiao, our Vice Chairman and Chief Technical Officer, under the HSR Act relating to her purchases of Common Stock.
In February 2019, Dr. Phillip Frost, our Chairman and Chief Executive Officer, paid a filing fee of $280,000 to the FTC under the HSR Act in connection with filings made by us and Dr. Frost, relating to his purchases of Common Stock. We reimbursed Dr. Frost for the HSR filing fee.
On November 8, 2018, we entered into a credit agreement with an affiliate of Dr. Frost, pursuant to which the lender committed to provide us with an unsecured line of credit in the amount of $60 million. Borrowings under this line of credit bore interest at a rate of 10 % per annum and could have been repaid and reborrowed at any time. The credit agreement included various customary remedies for the lender following an event of default, including the acceleration of repayment of outstanding amounts under this line of credit. This line of credit would have matured on November 8, 2023. We repaid approximately $28.8 million that was borrowed in 2019 and terminated this line of credit on or around February 20, 2019.
We hold investments in Zebra (ownership 29 %), Neovasc (1 %), ChromaDex Corporation (0.1 %), COCP (3 %), NIMS (1 %), Eloxx (2 %), BioCardia (1 %) and LeaderMed Health Group Limited (47 %). These investments were considered related party transactions as a result of our executive management’s ownership interests and/or board representation in these entities. See further discussion of our investments in Note 5.
In the first quarter of 2019, we exercised Neovasc’s Series C warrants for $1.2 million and exchanged the Series A warrants and received a total of 22,660 additional shares of Neovasc common stock.
In November 2016, we entered into a Pledge Agreement with the Museum of Science, Inc. and the Museum of Science Endowment Fund, Inc. pursuant to which we contributed an aggregate of $1.0 million over a four-year period for constructing, equipping and the general operation of the Frost Science Museum. Dr. Frost and Mr. Richard Pfenniger serve on the Board of Trustees of the Frost Science Museum and Mr. Pfenniger is the Vice Chairman of the Board of Trustees.
We lease office space from Frost Real Estate Holdings, LLC (“Frost Holdings”) in Miami, Florida, where our principal executive offices are located. Effective August 1, 2019, we entered into an amendment to our lease agreement with Frost Holdings. The lease, as amended, is for approximately 29,500 square feet of space. The lease provides for payments of approximately $89 thousand per month in the first year increasing annually to $101 thousand per month in the fifth year, plus applicable sales tax. The rent is inclusive of operating expenses, property taxes and parking.
BioReference purchases and uses certain products acquired from InCellDx, a company in which we hold a 29 % minority interest.
121
Note 13 Employee Benefit Plans
Note 14 Commitments and Contingencies
In connection with our acquisitions of CURNA, OPKO Diagnostics and OPKO Renal, we agreed to pay future consideration to the sellers upon the achievement of certain events. As a result, as of December 31, 2021, we recorded $2.8 million as contingent consideration, with $0.5 million recorded within Accrued expenses and $2.3 million recorded within Other long-term liabilities in the accompanying Consolidated Balance Sheets. Refer to Note 6.
On March 1, 2019, the Company received a Civil Investigative Demand (“CID”) from the U.S. Department of Justice, Washington, DC. The CID sets forth document requests and interrogatories in connection with allegations that the Company and certain of its affiliates violated the False Claims Act and/or the Anti-Kickback Statute. On January 13, 2022, the Federal Government notified the U.S.D.C., Middle District Florida, Jacksonville Division, that it is declining to intervene in the matter but retains the right, via the Attorney General, to consent to any proposed dismals of the action by the Court. On February 9, 2022, the States of Florida, Georgia, and Commonwealth of Massachusetts notified the U.S.D.C., Middle District Florida, Jacksonville Division, that they are declining to intervene in the matter. Notwithstanding the above declinations, on February 17, 2022, the Company was served with the Relator’s Summons and Complaint (“Complaint”), which had been previously sealed. The complaint alleges violations of the False Claims Act, the California Fraud Preventions Act, the Florida False Claims Act, the Massachusetts False Claims Act, the Georgia False Medicaid Claims Act, and illegal kickbacks. The Company is reviewing and assessing the allegations made in the Complaint and, at this point, has not determined whether there is any merit to these claims nor can it determine the extent of any potential liability. While management cannot predict the outcome of these matters at this time, the ultimate outcome could be material to our business, financial condition, results of operations, and cash flows.
As previously reported, BioReference receives and is routinely required to respond to Civil Investigative Demands (“CID”) in the ordinary course of business. On November 26, 2019, BioReference received a CID from the U.S. Department of Justice (“DOJ”). The CID states that DOJ is investigating whether BioReference paid unlawful remuneration to health care practitioners in violation of the Anti-Kickback Statute or Stark law and thus submitted or caused to be submitted false claims to government health care programs in violation of the False Claims Act. The time period covered by DOJ’s requests is January 1, 2011 through November 26, 2019. BioReference has fully cooperated with the DOJ by submitting the requested information and making current employees available for interviews, and DOJ recently made a presentation to BioReference regarding its position. The parties have reached verbal agreement on the settlement amount, which is anticipated to be approximately $10 million, excluding attorney fees.
On April 8, 2019, MabVax Therapeutics Holdings, Inc. filed a lawsuit in the Superior Court of California, County of San Diego against a number of individuals and entities, including the Company, Dr. Frost, Steven Rubin, the Company’s Executive Vice President-Administration, and an entity affiliated with Dr. Frost, based on the allegations raised in the SEC Complaint. The lawsuit seeks an award for actual and punitive damages, pre- and post-judgment interest; that the defendants be required to make full disclosure and accounting of their interests and transactions in plaintiff’s securities; costs of the suit, and reasonable attorney’s fees; and such other legal and equitable relief as the Court may deem proper under the circumstances. On January 31, 2022, plaintiffs entered into a confidential mutual release and settlement agreement with the Company, Dr. Frost, Frost Gamma Investment Trust, and Steve Rubin (the “Settlement Agreement”). The Settlement Agreement is subject to the approval of United States Bankruptcy Court for the District of Delaware.
On April 5, 2019, former shareholders of Claros Diagnostics, Inc. filed a complaint in the Chancery Court of Delaware against the Company, alleging among other things, that the Company breached the Agreement and Plan of Merger dated October 13, 2011 by and among the Company, Claros Merger Subsidiary, LLC and Claros Diagnostics, Inc. (the “Claros Merger Agreement”): (i) by failing to make a milestone payment of $2.375 million (payable in OPKO Common Stock) upon obtaining FDA approval of the Claros PSA test; and (ii) by repudiating its obligations to make additional future milestone payments as required under the Claros Merger Agreement. In January 2021, the Company and the shareholder representative entered into a settlement agreement providing, among other things, that the Company pay the shareholders $1.2 million, which the Company has paid in full.
In April 2017, the Civil Division of the United States Attorney’s Office for the Southern District of New York (the “SDNY”) informed BioReference that it believed that, from 2008 to 2012, BioReference had, in violation of the False Claims Act, improperly billed Medicare and TRICARE (both are federal government healthcare programs) for clinical laboratory
122
services provided to hospital inpatient beneficiaries at certain hospitals. In April 2019, the SDNY also informed BioReference that it believed that BioReference provided physicians subsidies for electronic health record systems prior to 2012 that violated regulations adopted by HHS in 2006 which allowed laboratories to provide these donations under certain conditions. BioReference and the SDNY reached a settlement with respect to these matters and a final settlement and release, including BioReference’s payment of an approximately $11.5 million settlement amount, was approved on September 22, 2020. The amount of related attorneys’ fees is currently being negotiated.
From time to time, we may receive inquiries, document requests, CIDs or subpoenas from the Department of Justice, OCR, CMS, various payors and fiscal intermediaries, and other state and federal regulators regarding investigations, audits and reviews. In addition to the matters discussed in this note, we are currently responding to CIDs, subpoenas, payor audits, and document requests for various matters relating to our laboratory operations. Some pending or threatened proceedings against us may involve potentially substantial amounts as well as the possibility of civil, criminal, or administrative fines, penalties, or other sanctions, which could be material. Settlements of suits involving the types of issues that we routinely confront may require monetary payments as well as corporate integrity agreements. Additionally, qui tam or “whistleblower” actions initiated under the civil False Claims Act may be pending but placed under seal by the court to comply with the False Claims Act’s requirements for filing such suits. Also, from time to time, we may detect issues of non-compliance with federal healthcare laws pertaining to claims submission and reimbursement practices and/or financial relationships with physicians, among other things. We may avail ourselves of various mechanisms to address these issues, including participation in voluntary disclosure protocols. Participating in voluntary disclosure protocols can have the potential for significant settlement obligations or even enforcement action. The Company generally has cooperated, and intends to continue to cooperate, with appropriate regulatory authorities as and when investigations, audits and inquiries arise.
We are a party to other litigation in the ordinary course of business. While we cannot predict the ultimate outcome of legal matters, we accrue a liability for legal contingencies when we believe that it is both probable that a liability has been incurred and that we can reasonably estimate the amount of the loss. It’s reasonably possible the ultimate liability could exceed amounts currently estimated and we review established accruals and adjust them to reflect ongoing negotiations, settlements, rulings, advice of legal counsel and other relevant information. To the extent new information is obtained and our views on the probable outcomes of claims, suits, assessments, investigations or legal proceedings change, changes in our accrued liabilities would be recorded in the period in which such determination is made. Because of the high degree of judgment involved in establishing loss estimates, the ultimate outcome of such matters will differ from our estimates and such differences may be material to our business, financial condition, results of operations, and cash flows.
Note 15 Revenue Recognition
We generate revenues from services, products and intellectual property as follows:
Revenue from services
Revenue for laboratory services is recognized at the time test results are reported, which approximates when services are provided and the performance obligations are satisfied. Services are provided to patients covered by various third-party payor programs including various managed care organizations, as well as the Medicare and Medicaid programs. Billings for services are included in revenue net of allowances for contractual discounts, allowances for differences between the amounts billed and estimated program payment amounts, and implicit price concessions provided to uninsured patients which are all elements of variable consideration.
The following are descriptions of our payors for laboratory services:
Healthcare Insurers. Reimbursements from healthcare insurers are based on negotiated fee-for-service schedules. Revenues consist of amounts billed, net of contractual allowances for differences between amounts billed and the estimated consideration we expect to receive from such payors, which considers historical denial and collection experience and the terms of our contractual arrangements. Adjustments to the allowances, based on actual receipts from the third-party payors, are recorded upon settlement.
Government Payors. Reimbursements from government payors are based on fee-for-service schedules set by governmental authorities, including traditional Medicare and Medicaid. Revenues consist of amounts billed, net of contractual allowances for differences between amounts billed and the estimated consideration we expect to receive from such payors,
123
which considers historical denial and collection experience and the terms of our contractual arrangements. Adjustments to the allowances, based on actual receipts from the government payors, are recorded upon settlement.
Client Payors. Client payors include physicians, hospitals, employers, and other institutions for which services are performed on a wholesale basis, and are billed and recognized as revenue based on negotiated fee schedules. Client payers also include cities, states and companies for which BioReference provides COVID-19 testing services.
Patients. Uninsured patients are billed based on established patient fee schedules or fees negotiated with physicians on behalf of their patients. Insured patients (including amounts for coinsurance and deductible responsibilities) are billed based on fees negotiated with healthcare insurers. Collection of billings from patients is subject to credit risk and ability of the patients to pay. Revenues consist of amounts billed net of discounts provided to uninsured patients in accordance with our policies and implicit price concessions. Implicit price concessions represent differences between amounts billed and the estimated consideration that we expect to receive from patients, which considers historical collection experience and other factors including current market conditions. Adjustments to the estimated allowances, based on actual receipts from the patients, are recorded upon settlement.
The complexities and ambiguities of billing, reimbursement regulations and claims processing, as well as considerations unique to Medicare and Medicaid programs, require us to estimate the potential for retroactive adjustments as an element of variable consideration in the recognition of revenue in the period the related services are rendered. Actual amounts are adjusted in the period those adjustments become known. For the years ended December 31, 2021, and December 31, 2020, positive revenue adjustments due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $40.4 million and $0.3 million were recognized, respectively. For the years ended December 31, 2019, revenue reductions due to changes in estimates of implicit price concessions for performance obligations satisfied in prior periods of $24.8 million were recognized.
Third-party payors, including government programs, may decide to deny payment or recoup payments for testing they contend were improperly billed or not medically necessary, against their coverage determinations, or for which they believe they have otherwise overpaid (including as a result of their own error), and we may be required to refund payments already received. Our revenues may be subject to retroactive adjustment as a result of these factors among others, including without limitation, differing interpretations of billing and coding guidance and changes by government agencies and payors in interpretations, requirements, and “conditions of participation” in various programs. We have processed requests for recoupment from third-party payors in the ordinary course of our business, and it is likely that we will continue to do so in the future. If a third-party payer denies payment for testing or recoups money from us in a later period, reimbursement for our testing could decline.
As an integral part of our billing compliance program, we periodically assess our billing and coding practices, respond to payor audits on a routine basis, and investigate reported failures or suspected failures to comply with federal and state healthcare reimbursement requirements, as well as overpayment claims which may arise from time to time without fault on the part of the Company. We may have an obligation to reimburse Medicare, Medicaid, and third-party payors for overpayments regardless of fault. We have periodically identified and reported overpayments, reimbursed payors for overpayments and taken appropriate corrective action.
Settlements with third-party payors for retroactive adjustments due to audits, reviews or investigations are also considered variable consideration and are included in the determination of the estimated transaction price for providing services. These settlements are estimated based on the terms of the payment agreement with the payor, correspondence from the payor and our historical settlement activity, including an assessment of the probability a significant reversal of cumulative revenue recognized will occur when the uncertainty is subsequently resolved. Estimated settlements are adjusted in future periods as adjustments become known (that is, new information becomes available), or as years are settled or are no longer subject to such audits, reviews, and investigations. As of December 31, 2021 and 2020, we have liabilities of approximately $5.0 million and $14.9 million within Accrued expenses and Other long-term liabilities related to reimbursements for payor overpayments.
124
The composition of Revenue from services by payor for the years ended December 31, 2021, 2020 and 2019 is as follows:
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Healthcare insurers | $ | $ | $ | ||||||||||||||
| Government payors | |||||||||||||||||
| Client payors | |||||||||||||||||
| Patients | |||||||||||||||||
| Total | $ | $ | $ | ||||||||||||||
Revenue from products
We recognize revenue from product sales when a customer obtains control of promised goods or services. The amount of revenue that is recorded reflects the consideration that we expect to receive in exchange for those goods or services. Our estimates for sales returns and allowances are based upon the historical patterns of product returns and allowances taken, matched against the sales from which they originated, and our evaluation of specific factors that may increase or decrease the risk of product returns. Product revenues are recorded net of estimated rebates, chargebacks, discounts, co-pay assistance and other deductions (collectively, “Sales Deductions”) as well as estimated product returns which are all elements of variable consideration. Allowances are recorded as a reduction of revenue at the time product revenues are recognized. The actual amounts of consideration ultimately received may differ from our estimates. If actual results in the future vary from our estimates, we will adjust these estimates, which would affect Revenue from products in the period such variances become known.
Rayaldee is distributed in the U.S. principally through the retail pharmacy channel, which initiates with the largest wholesalers in the U.S. (collectively, “Rayaldee Customers”). In addition to distribution agreements with Rayaldee Customers, we have entered into arrangements with many healthcare providers and payors that provide for government-mandated and/or privately-negotiated rebates, chargebacks and discounts with respect to the purchase of Rayaldee.
We recognize revenue for shipments of Rayaldee at the time of delivery to customers after estimating Sales Deductions and product returns as elements of variable consideration utilizing historical information and market research projections. For the years ended December 31, 2021, 2020 and 2019, we recognized $27.0 million, $36.8 million and $31.4 million in net product revenue from sales of Rayaldee.
The following table presents an analysis of product sales allowances and accruals as contract liabilities for the years ended December 31, 2021, 2020 and 2019:
| (In thousands) | Chargebacks, discounts, rebates and fees | Governmental | Returns | Total | ||||||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||
| Provision related to current period sales | ||||||||||||||||||||||||||
| Credits or payments made | ( |
( |
( |
( |
||||||||||||||||||||||
| Balance at December 31, 2021 | $ | $ | $ | $ | ||||||||||||||||||||||
Total gross Rayaldee sales
|
$ | |||||||||||||||||||||||||
Provision for Rayaldee sales allowances and accruals as a percentage of gross Rayaldee sales
|
% | |||||||||||||||||||||||||
125
| (In thousands) | Chargebacks, discounts, rebates and fees | Governmental | Returns | Total | ||||||||||||||||||||||
| Balance at December 31, 2019 | $ | $ | $ | $ | ||||||||||||||||||||||
| Provision related to current period sales | ||||||||||||||||||||||||||
| Credits or payments made | ( |
( |
( |
( |
||||||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | $ | $ | ||||||||||||||||||||||
Total gross Rayaldee sales
|
$ | |||||||||||||||||||||||||
Provision for Rayaldee sales allowances and accruals as a percentage of gross Rayaldee sales
|
% | |||||||||||||||||||||||||
| (In thousands) | Chargebacks, discounts, rebates and fees | Governmental | Returns | Total | ||||||||||||||||||||||
| Balance at December 31, 2018 | $ | $ | $ | $ | ||||||||||||||||||||||
| Provision related to current period sales | ||||||||||||||||||||||||||
| Credits or payments made | ( |
( |
( |
( |
||||||||||||||||||||||
| Balance at December 31, 2019 | $ | $ | $ | $ | ||||||||||||||||||||||
Total gross Rayaldee sales
|
$ | |||||||||||||||||||||||||
Provision for Rayaldee sales allowances and accruals as a percentage of gross Rayaldee sales
|
% | |||||||||||||||||||||||||
Taxes collected from customers related to revenues from services and revenues from products are excluded from revenues.
Revenue from intellectual property
We recognize revenues from the transfer of intellectual property generated through license, development, collaboration and/or commercialization agreements. The terms of these agreements typically include payment to us for one or more of the following: non-refundable, up-front license fees; development and commercialization milestone payments; funding of research and/or development activities; and royalties on sales of licensed products. Revenue is recognized upon satisfaction of a performance obligation by transferring control of a good or service to the customer.
For research, development and/or commercialization agreements that result in revenues, we identify all material performance obligations, which may include a license to intellectual property and know-how, and research and development activities. In order to determine the transaction price, in addition to any upfront payment, we estimate the amount of variable consideration at the outset of the contract either utilizing the expected value or most likely amount method, depending on the facts and circumstances relative to the contract. We constrain (reduce) our estimates of variable consideration such that it is probable that a significant reversal of previously recognized revenue will not occur throughout the life of the contract. When determining if variable consideration should be constrained, we consider whether there are factors outside of our control that could result in a significant reversal of revenue. In making these assessments, we consider the likelihood and magnitude of a potential reversal of revenue. These estimates are re-assessed each reporting period as required.
Upfront License Fees: If a license to our intellectual property is determined to be functional intellectual property distinct from the other performance obligations identified in the arrangement, we recognize revenue from nonrefundable, upfront license fees based on the relative value prescribed to the license compared to the total value of the arrangement. The revenue is recognized when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are not distinct from other obligations identified in the arrangement, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If the combined performance obligation is satisfied over time, we apply an appropriate method of measuring progress for purposes of recognizing revenue from nonrefundable, upfront license fees. We evaluate the measure of progress each reporting period and, if necessary, adjust the measure of performance and related revenue recognition.
126
Development and Regulatory Milestone Payments: Depending on facts and circumstances, we may conclude that it is appropriate to include the milestone in the estimated transaction price or that it is appropriate to fully constrain the milestone. A milestone payment is included in the transaction price in the reporting period that we conclude that it is probable that recording revenue in the period will not result in a significant reversal in amounts recognized in future periods. We may record revenues from certain milestones in a reporting period before the milestone is achieved if we conclude that achievement of the milestone is probable and that recognition of revenue related to the milestone will not result in a significant reversal in amounts recognized in future periods. We record a corresponding contract asset when this conclusion is reached. Milestone payments that have been fully constrained are not included in the transaction price to date. These milestones remain fully constrained until we conclude that achievement of the milestone is probable and that recognition of revenue related to the milestone will not result in a significant reversal in amounts recognized in future periods. We re-evaluate the probability of achievement of such development milestones and any related constraint each reporting period. We adjust our estimate of the overall transaction price, including the amount of revenue recorded, if necessary.
Research and Development Activities: If we are entitled to reimbursement from our customers for specified research and development expenses, we account for them as separate performance obligations if distinct. We also determine whether the research and development funding would result in revenues or an offset to research and development expenses in accordance with provisions of gross or net revenue presentation. The corresponding revenues or offset to research and development expenses are recognized as the related performance obligations are satisfied.
Sales-based Milestone and Royalty Payments: Our customers may be required to pay us sales-based milestone payments or royalties on future sales of commercial products. We recognize revenues related to sales-based milestone and royalty payments upon the later to occur of (i) achievement of the customer’s underlying sales or (ii) satisfaction of any performance obligation(s) related to these sales, in each case assuming the license to our intellectual property is deemed to be the predominant item to which the sales-based milestones and/or royalties relate.
Other Potential Products and Services: Arrangements may include an option for license rights, future supply of drug substance or drug product for either clinical development or commercial supply at the licensee’s election. We assess if these options provide a material right to the licensee and if so, they are accounted for as separate performance obligations at the inception of the contract and revenue is recognized only if the option is exercised and products or services are subsequently delivered or when the rights expire. If the promise is based on market terms and not considered a material right, the option is accounted for if and when exercised. If we are entitled to additional payments when the licensee exercises these options, any additional payments are generally recorded in license or other revenues when the licensee obtains control of the goods, which is upon delivery.
For the years ended December 31, 2021, 2020 and 2019 we recorded $25.8 million, $53.2 million and $73.3 million of revenue from the transfer of intellectual property and other, respectively. For the year ended December 31, 2021, revenue from transfer of intellectual property and other principally reflects $10.8 million of revenue related to the Pfizer Transaction, $1.0 million related to the LeaderMed joint venture (as defined below), $4.9 million related to the CAMP4 Agreement (as defined below) and a $5.0 million non-refundable upfront payment received under the Nicoya Agreement (as defined below). For the years ended December 31, 2020, and 2019 revenue from transfer of intellectual property and other principally reflects $28.7 million and $66.8 million of revenue related to the Pfizer Transaction. In addition, revenue from the transfer of intellectual property and other for the year ended December 31, 2020 included $16.2 million of grants received by BioReference under the CARES Act and a $3 million milestone payment triggered by the first marketing approval of Rayaldee in Europe.
Contract liabilities relate to cash consideration that OPKO receives in advance of satisfying the related performance obligations. Changes in the contractual liabilities balance for the year ended December 31, 2021 are as follows:
| (In thousands) | ||||||||
| Balance at December 31, 2020 | $ | |||||||
| Balance at December 31, 2021 | ||||||||
| Revenue recognized in the period from: | ||||||||
| Amounts included in contracts liability at the beginning of the period | $ | |||||||
The contract liability balance at December 31, 2021 related primarily to accelerated payments received as part of the CARES Act. Refer to Note 2.
Note 16 Strategic Alliances
127
LeaderMed
On September 14, 2021, we and LeaderMed Health Group Limited (“LeaderMed”), a pharmaceutical development company with operations based in Asia, announced the formation of a joint venture to develop, manufacture and commercialize two of OPKO’s clinical stage, long-acting drug products in Greater China and eight other Asian territories.
Under the terms of the agreements, we have granted the joint venture exclusive rights to develop, manufacture and commercialize (a) OPK88003, an oxyntomodulin analog being developed for the treatment of obesity and diabetes, and (b) Factor VIIa-CTP, a novel long-acting coagulation factor being developed to treat hemophilia, in exchange for a 47 % ownership interest in the joint venture. In addition, we received an upfront payment of $1 million and will be reimbursed for clinical trial material and technical support we provide the joint venture. For the year ended December 31, 2021, we recognized the upfront payment of $1 million as revenue from transfer of intellectual property and other.
LeaderMed has agreed to be responsible for funding the joint venture’s operations, development and commercialization efforts and, together with its syndicate partners, initially invested $11 million in exchange for a 53 % ownership interest. We retain full rights to oxyntomodulin and Factor VIIa-CTP in all other geographies.
CAMP4 Therapeutics
On July 6, 2021, we entered into an exclusive license agreement (the “CAMP4 Agreement”) with CAMP4, pursuant to which we granted to CAMP4 an exclusive license to develop, manufacture, commercialize or improve therapeutics utilizing the AntagoNAT technology, an oligonucleotide platform developed under OPKO CURNA, which includes the molecule for the treatment of Dravet syndrome, together with any derivative or modification thereof (the “Licensed Compound”) and any pharmaceutical product that comprises or contains the Licensed Compound, alone or in combination with one or more other active ingredients (“Licensed Product”), worldwide. The CAMP4 Agreement grant covers human pharmaceutical, prophylactic, and therapeutic and certain diagnostic uses.
We received an initial upfront payment of $1.5 million and 3,373,008 shares of CAMP4’s Series A Prime Preferred Stock (“Preferred Stock”), which equates to approximately 9 % of the outstanding shares of CAMP4, and we are eligible to receive up to $3.5 million in development milestone payments for Dravet syndrome products, and $4 million for non-Dravet syndrome products, as well as sales milestones of up to $90 million for Dravet syndrome products and up to $90 million for non-Dravet syndrome products. We may also receive double digit royalty payments on the net sales of royalty bearing products, subject to adjustment. In addition, upon achievement of certain development milestones, we will be eligible to receive equity consideration of up to 5,782,299 shares of Preferred Stock in connection with Dravet syndrome products and up to 1,082,248 shares of Preferred Stock in connection with non-Dravet syndrome products. In connection with our acquisition of CURNA, we agreed to pay future consideration to the sellers upon the achievement of certain events. As a result of our execution of the CAMP4 Agreement, we will have to pay a percentage of any payments received under the CAMP4 Agreement to the former CURNA stockholders. For the three months ended September 30, 2021, we recognized the fair value of the upfront payments of cash and shares of Preferred Stock totaling $4.9 million in revenue from transfer of intellectual property and other.
Unless earlier terminated, the CAMP4 Agreement will remain in effect on a Licensed Product-by-Licensed Product and country by-country basis until such time as the royalty term expires for a Licensed Product in a country, and expires in its entirety upon the expiration of the royalty term for the last Licensed Product in the last country. CAMP4’s royalty obligations expire on the later of (i) the expiration, invalidation or abandonment date of the last patent right in connection with the royalty bearing product, or (ii) ten (10 ) years after a royalty bearing product’s first commercial sale in a country. In addition to termination rights for material breach and bankruptcy, CAMP4 is permitted to terminate the Agreement after a specified notice period.
NICOYA Macau Limited
On June 18, 2021, EirGen, our wholly owned subsidiary, and NICOYA Macau Limited (“Nicoya”), a Macau corporation and an affiliate of NICOYA Therapeutics, entered into a Development and License Agreement (the “Nicoya Agreement”) granting Nicoya the exclusive rights for the development and commercialization of extended release calcifediol (the “Nicoya Product”) in Greater China, which includes mainland China, Hong Kong, Macau, and Taiwan (collectively, the “Nicoya Territory”). Extended release calcifediol is marketed in the U.S. by OPKO under the tradename Rayaldee. The license grant to Nicoya covers the therapeutic and preventative use of the Nicoya Product for SHPT in non-dialysis and hemodialysis chronic kidney disease patients (the “Nicoya Field”).
EirGen has received an initial upfront payment of $5 million and is eligible to receive an additional $5 million upon the first to occur of (A) a predetermined milestone and (B) the first anniversary of the effective date. EirGen is also eligible to receive up to an additional aggregate amount of $115 million upon the achievement of certain development, regulatory and
128
sales-based milestones by Nicoya for the Nicoya Product in the Nicoya Territory. EirGen will also receive tiered, double digit royalty payments at rates in the low double digits on net product sales within the Nicoya Territory and in the Nicoya Field.
Nicoya will, at its sole cost and expense, be responsible for performing all development activities necessary to obtain all regulatory approvals for the Nicoya Product in the Nicoya Territory and for all commercial activities pertaining to the Nicoya Product in the Nicoya Territory.
Unless earlier terminated, the Nicoya Agreement will remain in effect until such time as all royalty payment terms and extended payment terms have expired, and Nicoya shall have no further payment obligations to EirGen under the terms of the Nicoya Agreement. Nicoya’s royalty obligations expire on the later of (i) expiration of the last to expire valid patent claim covering the Nicoya Product sold in the Nicoya Territory, (ii) expiration of all regulatory and data exclusivity applicable to the Nicoya Product in the Nicoya Territory, and (iii) on a product-by-product basis, ten (10 ) years after such Nicoya Product’s first commercial sale in the Nicoya Territory. In addition to termination rights for material breach and bankruptcy, Nicoya is permitted to terminate the Nicoya Agreement after a specified notice period.
Vifor Fresenius Medical Care Renal Pharma Ltd
In May 2016, EirGen and Vifor Fresenius Medical Care Renal Pharma Ltd (“VFMCRP”), entered into a Development and License Agreement (the “VFMCRP Agreement”) for the development and commercialization of Rayaldee (the “Product”) worldwide, except for (i) the U.S., (ii) any country in Central America or South America (excluding Mexico), (iii) Russia, (iv) China, (v) Japan, (vi) Ukraine, (vii) Belorussia, (viii) Azerbaijan, (ix) Kazakhstan, and (x) Taiwan (the “VFMCRP Territory”). The license to VFMCRP potentially covers all therapeutic and prophylactic uses of the Product in human patients (the “VFMCRP Field”), provided that initially the license is for the use of the Product for the treatment or prevention of SHPT related to patients with CKD and vitamin D insufficiency/deficiency (the “VFMCRP Initial Indication”).
Effective May 23, 2021, we entered into an amendment to the VFMCRP Agreement pursuant to which the parties thereto agreed to include Japan as part of the VFMCRP Territory.
Effective May 5, 2020, we entered into an amendment to the VFMCRP Agreement pursuant to which the parties agreed to exclude Mexico, South Korea, the Middle East and all of the countries of Africa from the VFMCRP Territory. In addition, the parties agreed to certain amendments to the milestone structure and to reduce minimum royalties payable. As revised, the Company has received a $3 million payment triggered by the first marketing approval of Rayaldee in Europe and is eligible to receive up to an additional $17 million in regulatory milestones and $210 million in milestone payments tied to launch, pricing and sales of Rayaldee, and tiered, double-digit royalties.
We plan to share responsibility with VFMCRP for the conduct of trials specified within an agreed-upon development plan, with each company leading certain activities within the plan. EirGen will lead the manufacturing activities within and outside the VFMCRP Territory and the commercialization activities outside the VFMCRP Territory and outside the VFMCRP Field in the VFMCRP Territory and VFMCRP will lead the commercialization activities in the VFMCRP Territory and the VFMCRP Field. For the initial development plan, the companies have agreed to certain cost sharing arrangements. VFMCRP will be responsible for all other development costs that VFMCRP considers necessary to develop the Product for the use of the Product for the VFMCRP Initial Indication in the VFMCRP Territory in the VFMCRP Field except as otherwise provided in the VFMCRP Agreement. The first of the clinical studies provided for in the development activities commenced in September 2018.
In connection with the VFMCRP Agreement, the parties entered into a letter agreement pursuant to which EirGen granted to VFMCRP an exclusive option (the “Option”) to acquire an exclusive license under certain EirGen patents and technology to use, import, offer for sale, sell, distribute and commercialize the Product in the U.S. solely for the treatment of SHPT in dialysis patients with CKD and vitamin D insufficiency (the “Dialysis Indication”). Upon exercise of the Option, VFMCRP will reimburse EirGen for all of the development costs incurred by EirGen with respect to the Product for the Dialysis Indication in the U.S. VFMCRP would also pay EirGen up to an additional aggregate amount of $555 million of sales-based milestones upon the achievement of certain milestones and would be obligated to pay royalties at percentage rates that range from the mid-teens to the mid-twenties on sales of the Product in the U.S. for the Dialysis Indication. To date, VFMCRP has not exercised its option.
Payments received for Regulatory Milestones and Sales Milestones are non-refundable. The Regulatory Milestones are payable if and when VFMCRP obtains approval from certain regulatory authorities and will be recognized as revenue in the period in which the associated milestone is achieved, assuming all other revenue recognition criteria are met. We account for the Sales Milestones as royalties and Sales Milestones payments will be recognized as revenue in the period in which the associated milestone is achieved or sales occur, assuming all other revenue recognition criteria are met.
129
Pfizer Inc.
In December 2014, we entered into an exclusive worldwide agreement (the “Pfizer Agreement”) with Pfizer for the development and commercialization of our long-acting Somatrogon (hGH-CTP) for the treatment of growth hormone deficiency (“GHD”) in adults and children, as well as for the treatment of growth failure in children born small for gestational age (the “Pfizer Transaction”).
In early 2022, the European Commission and Ministry of Health, Labour and Welfare in Japan approved the next-generation long-acting recombinant human growth hormone NGENLA (Somatrogon), a once-weekly injection to treat pediatric growth hormone deficiency. Further, Canada and Australia approved NGENLA in October and November of 2021, respectively.
In January 2022, the FDA issued a Complete Response Letter for the BLA for Somatrogon. Pfizer and OPKO are evaluating the FDA’s comments and will work with the agency to determine the best path forward for Somatrogon (hGH-CTP) in the United States.
In May 2020, we entered into an Amended and Restated Development and Commercialization License Agreement (the “Restated Pfizer Agreement”) with Pfizer, effective January 1, 2020, pursuant to which the parties agreed, among other things, to share all costs for Manufacturing Activities, as defined in the Restated Pfizer Agreement, for developing a licensed product for the three indications included in the Restated Pfizer Agreement.
On October 21, 2019, we and Pfizer announced that the global phase 3 trial evaluating Somatrogon dosed once-weekly in prepubertal children with GHD met its primary endpoint of non-inferiority to daily Genotropin® (somatropin) for injection, as measured by annual height velocity at 12 months.
Under the terms of the Pfizer Transaction, as restated, we received non-refundable and non-creditable upfront payments of $295.0 million and are eligible to receive up to an additional $275.0 million upon the achievement of certain regulatory milestones. Pfizer received the exclusive license to commercialize Somatrogon worldwide. In addition, we are eligible to receive initial tiered royalty payments associated with the commercialization of Somatrogon for adult GHD with percentage rates ranging from the high teens to mid-twenties. Upon the launch of Somatrogon for pediatric GHD in certain major markets, the royalties will transition to regional, tiered gross profit sharing for both Somatrogon and Pfizer’s Genotropin®.
The agreement with Pfizer will remain in effect until the last sale of the licensed product, unless earlier terminated as permitted under the Pfizer Agreement. In addition to termination rights for material breach and bankruptcy, Pfizer is permitted to terminate the Pfizer Agreement in its entirety, or with respect to one or more world regions, without cause after a specified notice period. If the Pfizer Agreement is terminated by us for Pfizer’s uncured material breach, or by Pfizer without cause, provision has been made for transition of product and product responsibilities to us for the terminated regions, as well as continued supply of product by Pfizer or transfer of supply to us in order to support the terminated regions.
We recognized the non-refundable $295.0 million upfront payments as revenue as the research and development services were completed and as of December 31, 2021 and 2020, we had no contract liabilities related to the Pfizer Transaction.
The Pfizer Transaction includes milestone payments of $275.0 million upon the achievement of certain milestones. The milestones range from $20.0 million to $90.0 million each and are based on achievement of regulatory approval in the U.S. and regulatory approval and price approval in other major markets. The milestone payments will be recognized as revenue in the period in which the associated milestone is achieved, assuming all other revenue recognition criteria are met. To date, no revenue has been recognized related to the achievement of the milestones.
Other
We have completed strategic deals with numerous institutions and commercial partners. In connection with these agreements, upon the achievement of certain milestones we are obligated to make certain payments and have royalty obligations upon sales of products developed under the license agreements. At this time, we are unable to estimate the timing and amounts of payments as the obligations are based on future development of the licensed products.
Note 17 Leases
130
over the term of the lease within a particular currency environment. We used the incremental borrowing rates as of January 1, 2019 for operating leases that commenced prior to that date. Many of our leases contain rental escalation, renewal options and/or termination options that are factored into our determination of lease payments as appropriate. Variable lease payment amounts that cannot be determined at the commencement of the lease are not included in the right-to-use assets or liabilities.
We elected the use of permitted practical expedients of not recording leases on our Consolidated Balance Sheet when the leases have terms of 12 months or less, and we elected not to separate nonlease components from lease components and instead account for each separate lease component and the nonlease components associated with that lease component as a single lease component.
The following table presents the lease balances within the Consolidated Balance Sheet as of December 31, 2021 and 2020:
| (in thousands) | Classification on the Balance Sheet | December 31, 2021 | December 31, 2020 | |||||||||||||||||
| Assets | ||||||||||||||||||||
| Operating lease assets | Operating lease right-of-use assets | $ | $ | |||||||||||||||||
| Property, plant and equipment, net | ||||||||||||||||||||
| Liabilities | ||||||||||||||||||||
| Current | ||||||||||||||||||||
| Operating lease liabilities | Current maturities of operating leases | |||||||||||||||||||
| Current maturities of finance leases | ||||||||||||||||||||
| Long-term | ||||||||||||||||||||
| Operating lease liabilities | Operating lease liabilities | |||||||||||||||||||
| Finance lease liabilities | $ | $ | ||||||||||||||||||
| Weighted average remaining lease term | ||||||||||||||||||||
| Operating leases | ||||||||||||||||||||
| Finance leases | ||||||||||||||||||||
| Weighted average discount rate | ||||||||||||||||||||
| Operating leases | % | % | ||||||||||||||||||
| Finance leases | % | % | ||||||||||||||||||
The following table reconciles the undiscounted future minimum lease payments (displayed by year and in the aggregate) under noncancelable operating leases with terms of more than one year to the total operating lease liabilities recognized on our Consolidated Balance Sheet as of December 31, 2021:
| (in thousands) | Operating | Finance | |||||||||
| 2022 | $ | $ | |||||||||
| 2023 | |||||||||||
| 2024 | |||||||||||
| 2025 | |||||||||||
| 2026 | |||||||||||
| Thereafter | |||||||||||
| Total undiscounted future minimum lease payments | |||||||||||
| Less: Difference between lease payments and discounted lease liabilities | |||||||||||
| Total lease liabilities | $ | $ | |||||||||
Expense under operating leases and finance leases was $18.0 million and $2.3 million, respectively, for the year ended December 31, 2021, which includes $2.5 million of variable lease costs. Expense under operating leases and finance leases was $17.8 million and $3.0 million, respectively, for the year ended December 31, 2020, and includes $3.0 million of variable lease
131
costs. Expense under operating leases and finance leases was $20.2 million and $3.1 million, respectively, for the year ended December 31, 2019, and includes $3.4 million of variable lease costs. Operating lease costs and finance lease costs are included within Operating loss in the Consolidated Statement of Operations. Short-term lease costs were not material.
Supplemental cash flow information is as follows:
| (in thousands) | For the years ended December 31, | ||||||||||
| 2021 | 2020 | ||||||||||
| Operating cash out flows from operating leases | $ | $ | |||||||||
| Operating cash out flows from finance leases | |||||||||||
| Financing cash out flows from finance leases | |||||||||||
| Total | $ | $ | |||||||||
Note 18 Segments
We manage our operations in two reportable segments, pharmaceuticals and diagnostics. The pharmaceuticals segment consists of our pharmaceutical operations in Chile, Mexico, Ireland, Israel and Spain, Rayaldee product sales and our pharmaceutical research and development. The diagnostics segment primarily consists of our clinical laboratory operations through BioReference and our point-of-care operations. There are no significant inter-segment sales. We evaluate the performance of each segment based on operating profit or loss. There is no inter-segment allocation of interest expense and income taxes.
132
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Revenue from services: | |||||||||||||||||
| Pharmaceutical | $ | $ | $ | ||||||||||||||
| Diagnostics | |||||||||||||||||
| Corporate | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
| Revenue from products: | |||||||||||||||||
| Pharmaceutical | $ | $ | $ | ||||||||||||||
| Diagnostics | |||||||||||||||||
| Corporate | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
| Revenue from transfer of intellectual property and other: | |||||||||||||||||
| Pharmaceutical | $ | $ | $ | ||||||||||||||
| Diagnostics | |||||||||||||||||
| Corporate | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
| Operating income (loss): | |||||||||||||||||
| Pharmaceutical | $ | ( |
$ | ( |
$ | ( |
|||||||||||
| Diagnostics | ( |
||||||||||||||||
| Corporate | ( |
( |
( |
||||||||||||||
| $ | $ | $ | ( |
||||||||||||||
| Depreciation and amortization: | |||||||||||||||||
| Pharmaceutical | $ | $ | $ | ||||||||||||||
| Diagnostics | |||||||||||||||||
| Corporate | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
| Loss from investment in investees: | |||||||||||||||||
| Pharmaceutical | $ | ( |
$ | ( |
$ | ( |
|||||||||||
| Diagnostics | |||||||||||||||||
| Corporate | |||||||||||||||||
| $ | ( |
$ | ( |
$ | ( |
||||||||||||
| Revenues: | |||||||||||||||||
| U.S. | $ | $ | $ | ||||||||||||||
| Ireland | |||||||||||||||||
| Chile | |||||||||||||||||
| Spain | |||||||||||||||||
| Israel | |||||||||||||||||
| Mexico | |||||||||||||||||
| Other | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
133
| (In thousands) | December 31, 2021 |
December 31, 2020 |
|||||||||
| Assets: | |||||||||||
| Pharmaceutical | $ | $ | |||||||||
| Diagnostics | |||||||||||
| Corporate | |||||||||||
| $ | $ | ||||||||||
| Goodwill: | |||||||||||
| Pharmaceutical | $ | $ | |||||||||
| Diagnostics | |||||||||||
| Corporate | |||||||||||
| $ | $ | ||||||||||
No customer represented more than 10% of our total consolidated revenue during the years ended December 31, 2021, 2020 and 2019. As of December 31, 2021 and 2020, no customer represented more than 10% of our accounts receivable balance.
The following table reconciles our Property, plant and equipment, net between U.S. and foreign jurisdictions:
| (In thousands) | December 31, 2021 | December 31, 2020 | |||||||||
| PP&E: | |||||||||||
| U.S. | $ | $ | |||||||||
| Foreign | |||||||||||
| Total | $ | $ | |||||||||
Note 19 Fair Value Measurements
We record fair values at an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement determined based on assumptions that market participants would use in pricing an asset or liability. We utilize a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers are: Level 1, defined as observable inputs such as quoted prices in active markets; Level 2, defined as inputs other than quoted prices in active markets that are either directly or indirectly observable; and Level 3, defined as unobservable inputs in which little or no market data exists, therefore requiring an entity to develop its own assumptions.
As of December 31, 2021, we have equity securities (refer to Note 5), forward foreign currency exchange contracts for inventory purchases (refer to Note 20) and contingent consideration related to the acquisitions of CURNA, OPKO Diagnostics and OPKO Renal that are required to be measured at fair value on a recurring basis. In addition, in connection with our investment and our consulting agreement with BioCardia, we record the related BioCardia options at fair value as well as the warrants from COCP, InCellDx, Inc., Xenetic and Phio.
134
Our financial assets and liabilities measured at fair value on a recurring basis are as follows:
| Fair value measurements as of December 31, 2021 | |||||||||||||||||||||||
| (In thousands) | Quoted prices in active markets for identical assets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) |
Total | |||||||||||||||||||
| Assets: | |||||||||||||||||||||||
| Equity securities | $ | $ | $ | $ | |||||||||||||||||||
| Common stock options/warrants | |||||||||||||||||||||||
| Forward contracts | |||||||||||||||||||||||
| Total assets | $ | $ | $ | $ | |||||||||||||||||||
| Liabilities: | |||||||||||||||||||||||
| Contingent consideration: | |||||||||||||||||||||||
| Total liabilities | $ | $ | $ | $ | |||||||||||||||||||
| Fair value measurements as of December 31, 2020 | |||||||||||||||||||||||
| (In thousands) | Quoted prices in active markets for identical assets (Level 1) |
Significant other observable inputs (Level 2) |
Significant unobservable inputs (Level 3) |
Total | |||||||||||||||||||
| Assets: | |||||||||||||||||||||||
| Equity securities | $ | $ | $ | $ | |||||||||||||||||||
| Common stock options/warrants | |||||||||||||||||||||||
| Total assets | $ | $ | $ | $ | |||||||||||||||||||
| Liabilities: | |||||||||||||||||||||||
| Forward contracts | $ | $ | $ | $ | |||||||||||||||||||
| Contingent consideration: | $ | $ | $ | $ | |||||||||||||||||||
| Total liabilities | $ | $ | $ | $ | |||||||||||||||||||
The carrying amount and estimated fair value of our 2025 Notes, as well as the applicable fair value hierarchy tiers, are contained in the table below. The fair value of the 2025 Notes is determined using inputs other than quoted prices in active markets that are directly observable.
| December 31, 2021 | |||||||||||||||||||||||||||||
| (In thousands) | Carrying Value |
Total Fair Value |
Level 1 | Level 2 | Level 3 | ||||||||||||||||||||||||
| 2025 Notes | $ | $ | $ | $ | $ | ||||||||||||||||||||||||
There have been no transfers between Level 1 and Level 2 and no transfers to or from Level 3 of the fair value hierarchy.
As of December 31, 2021 and 2020, the carrying value of our other financial instrument assets approximates their fair value due to their short-term nature or variable rate of interest.
135
The following tables reconcile the beginning and ending balances of our Level 3 assets and liabilities as of December 31, 2021 and 2020:
| December 31, 2021 | |||||
| (In thousands) | Contingent consideration |
||||
| Balance at December 31, 2020 | $ | ||||
| Change in fair value: | |||||
| Included in results of operations | ( |
||||
| Foreign currency impact | |||||
| Payments | ( |
||||
| Balance at December 31, 2021 | $ | ||||
| December 31, 2020 | |||||
| (In thousands) | Contingent consideration |
||||
| Balance at December 31, 2019 | $ | ||||
| Change in fair value | |||||
| Included in results of operations | ( |
||||
| Balance at December 31, 2020 | $ | ||||
The estimated fair values of our financial instruments have been determined by using available market information and what we believe to be appropriate valuation methodologies. We use the following methods and assumptions in estimating fair value:
Contingent consideration – We estimate the fair value of the contingent consideration utilizing a discounted cash flow model for the expected payments based on estimated timing and expected revenues. We use several discount rates depending on each type of contingent consideration related to OPKO Diagnostics, CURNA and OPKO Renal transactions. As of December 31, 2021, of the $2.8 million of contingent consideration, $0.5 million is recorded in Accrued expenses and $2.3 million is recorded in Other long-term liabilities. As of December 31, 2020, of the $5.7 million of contingent consideration, $1.2 million is recorded in Accrued expenses and $4.5 million is recorded in Other long-term liabilities. As a result of our execution of the CAMP4 Agreement (as defined in Note 16), we will have to pay a percentage of any payments received under the CAMP4 Agreement to the former CURNA stockholders.
Note 20 Derivative Contracts
The following table summarizes the fair values and the presentation of our derivative financial instruments in the Consolidated Balance Sheets:
| (In thousands) | Balance Sheet Component | December 31, 2021 | December 31, 2020 |
||||||||||||||
| Derivative financial instruments: | |||||||||||||||||
| Common stock options/warrants | Investments, net | $ | $ | ||||||||||||||
| Forward contracts | Unrealized gains on forward contracts are recorded in Other current assets and prepaid expenses. Unrealized (losses) on forward contracts are recorded in Accrued expenses. | $ | $ | ( |
|||||||||||||
We enter into foreign currency forward exchange contracts with respect to the risk of exposure to exchange rate differences arising from inventory purchases on letters of credit. Under these forward contracts, for any rate above or below the fixed rate, we receive or pay the difference between the spot rate and the fixed rate for the given amount at the settlement date.
136
To qualify the derivative instrument as a hedge, we are required to meet strict hedge effectiveness and contemporaneous documentation requirements at the initiation of the hedge and assess the hedge effectiveness on an ongoing basis over the life of the hedge. At December 31, 2021 and 2020, our derivative financial instruments do not meet the documentation requirements to be designated as hedges. Accordingly, we recognize the changes in Fair value of derivative instruments, net in our Consolidated Statement of Operations. The following table summarizes the losses and gains recorded for the years ended December 31, 2021, 2020 and 2019:
| For the years ended December 31, | |||||||||||||||||
| (In thousands) | 2021 | 2020 | 2019 | ||||||||||||||
| Derivative gain (loss): | |||||||||||||||||
| Common stock options/warrants | $ | ( |
$ | ( |
$ | ( |
|||||||||||
| Forward contracts | $ | $ | $ | ||||||||||||||
| Total | $ | $ | $ | ||||||||||||||
Note 21 Selected Quarterly Financial Data (Unaudited)
| For the 2021 Quarters Ended | |||||||||||||||||||||||
| (In thousands, except per share data) | March 31 | June 30 | September 30 | December 31 | |||||||||||||||||||
| Total revenues | $ | $ | $ | $ | |||||||||||||||||||
| Total costs and expenses | |||||||||||||||||||||||
| Net income (loss) | ( |
( |
|||||||||||||||||||||
| Earnings (loss) per share, basic and diluted | $ | $ | ( |
$ | $ | ( |
|||||||||||||||||
| For the 2020 Quarters Ended | |||||||||||||||||||||||
| (In thousands, except per share data) | March 31 | June 30 | September 30 | December 31 | |||||||||||||||||||
| Total revenues | $ | $ | $ | $ | |||||||||||||||||||
| Total costs and expenses | |||||||||||||||||||||||
| Net income (loss) | ( |
||||||||||||||||||||||
| Earnings (loss) per share, basic and diluted | $ | ( |
$ | $ | $ | ||||||||||||||||||
Note 22 Subsequent Events
In February 2022, the European Commission approved the next-generation long-acting recombinant human growth hormone NGENLA (Somatrogon), a once-weekly injection to treat children and adolescents from as young as 3 years of age with growth disturbance due to insufficient secretion of growth hormone.
In January 2022, Pfizer, Inc. and OPKO announced that the FDA issued a Complete Response Letter for the BLA for Somatrogon. Somatrogon is an investigational once-weekly long-acting recombinant human growth hormone for the treatment of GHD in pediatric patients. Pfizer is evaluating the FDA’s comments and will work with the agency to determine an appropriate path forward.
In January 2022, Pfizer, Inc. and OPKO announced that the long-acting growth hormone injection, NGENLA® (Somatrogon) Inj. 24 mg Pens and 60 mg Pens, has been approved by the Ministry of Health, Labour and Welfare in Japan for the treatment of GHD in pediatric patients. Somatrogon has also been approved in the Canada and Japan under the brand name NGENLA.
In January 2022, Sema4 and OPKO announced they have signed GeneDx Merger Agreement, pursuant to which Sema4 has agreed to acquire GeneDx, a leader in genomic testing and analysis, from OPKO, subject to satisfaction of customary closing conditions. The GeneDx Transaction is expected to close in the second quarter of 2022.
Under the terms of the agreement, Sema4 has agreed to acquire GeneDx for an upfront payment of $150 million in cash together with 80.0 million shares of Sema4 Common Stock, subject to a customary purchase price adjustment mechanism providing for a normalized level of working capital and that GeneDx be free of debt at closing of the GeneDx Transaction. Additionally, Sema4 agreed to pay OPKO up to an additional $150 million revenue-based milestones over the next two years (which may be paid in Sema4 Common Stock, cash or a combination thereof in Sema4’s discretion, subject to GeneDx
137
138
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, have evaluated the effectiveness of the Company’s disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act ) as of December 31, 2021. Our disclosure controls and procedures are designed to provide reasonable assurance that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the rules and forms of the Securities and Exchange Commission. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Based on this evaluation, management concluded that our disclosure controls and procedures were effective as of December 31, 2021.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined effective could provide only reasonable assurance with respect to financial statement preparation and presentation.
Our management conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2021, based on the framework in the Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “2013 Internal Control-Integrated Framework”). Based on our evaluation under the 2013 Internal Control-Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2021.
The effectiveness of the Company’s internal control over financial reporting as of December 31, 2021 has been audited by Ernst & Young LLP, our independent registered public accounting firm, who also audited our Consolidated Financial Statements included in this Annual Report on Form 10-K, as stated in their report which appears with our accompanying Consolidated Financial Statements.
Changes to the Company’s Internal Control Over Financial Reporting
There have been no changes to the Company’s internal control over financial reporting that occurred during quarter ended December 31, 2021 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
139
PART III
The information required in Items 10 (Directors, Executive Officers and Corporate Governance), Item 11 (Executive Compensation), Item 12 (Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters), Item 13 (Certain Relationships and Related Transactions, and Director Independence), and Item 14 (Principal Accounting Fees and Services) is incorporated by reference to the Company’s definitive proxy statement for the 2022 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December 31, 2021.
140
PART IV.
Item 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
| (a) | (1) | Financial Statements: See Part II, Item 8 of this report. | ||||||
| Schedule I - Condensed Financial Information of Registrant. Additionally, the financial statement schedule entitled “Schedule II – Valuation and Qualifying Accounts” has been omitted since the information required is included in the consolidated financial statements and notes thereto. Other schedules are omitted because they are not required. | ||||||||
| (2) | Exhibits: See Index to Exhibits below. | |||||||
INDEX TO EXHIBITS
141
142
| 101.SCH | XBRL Taxonomy Extension Schema Document | ||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | ||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | ||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | ||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | ||||
* Denotes management contract or compensatory plan or arrangement.
** Filed herewith.
+ Certain confidential material contained in the document has been omitted and filed separately with the Securities and Exchange Commission.
(1)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on April 2, 2007, and incorporated herein by reference.
(2)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 8, 2008 for the Company’s three-month period ended June 30, 2008, and incorporated herein by reference.
(3)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on September 24, 2009, and incorporated herein by reference.
(4)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2009 for the Company’s three-month period ended September 30, 2009, and incorporated herein by reference.
(5)Filed with the Company’s Quarterly Report on Form 10-Q/A filed with the Securities and Exchange Commission on July 5, 2011, and incorporated herein by reference.
(6)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2012 for the Company’s three-month period ended September 30, 2012, and incorporated herein by reference.
(7)Filed with the Company’s Annual Report on Form 10-K/A filed with the Securities and Exchange Commission on July 28, 2011.
(8)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 5, 2013, and incorporated herein by reference.
(9)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 12, 2013 for the Company’s three month period ended September 30, 2013, and incorporated herein by reference.
(10)Filed with the Company’s Definitive Proxy Statement on Schedule 14A filed with the Securities and Exchange Commission on March 25, 2016, and incorporated herein by reference.
(11)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 8, 2016 for the Company’s three month period ended June 30, 2016, and incorporated herein by reference.
(12)Filed with the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 1, 2018 and incorporated herein by reference.
(13)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on February 7, 2019 and incorporated herein by reference.
(14)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on June 21, 2019.
(15)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on October 29, 2019.
(16)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on July 31, 2020.
(17)Filed with the Company’s Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 18, 2021 and incorporated herein by reference.
143
(18)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on May 7, 2021.
(19)Filed with the Company’s Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on July 29, 2021.
(20)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on September 3, 2021
(21)Filed with the Company’s Current Report on Form 8-K filed with the Securities and Exchange Commission on January 18, 2022.
Item 16. FORM 10-K SUMMARY.
None.
144
Schedule I - Condensed Financial Information of Registrant
OPKO Health, Inc.
PARENT COMPANY CONDENSED BALANCE SHEETS
(In thousands, except share and per share data)
| December 31, | |||||||||||
| 2021 | 2020 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | 24,592 | $ | 3,740 | |||||||
| Other current assets and prepaid expenses | 2,620 | 2,433 | |||||||||
| Total current assets | 27,212 | 6,173 | |||||||||
| Investments | 1,870,743 | 1,901,104 | |||||||||
| Operating lease right-of-use assets | 3,030 | 3,958 | |||||||||
| Other assets | 1 | 25 | |||||||||
| Total assets | $ | 1,900,986 | $ | 1,911,260 | |||||||
| LIABILITIES AND EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | 317 | $ | 1,194 | |||||||
| Accrued expenses | 21,484 | 8,324 | |||||||||
| Current maturities of operating leases | 1,203 | 1,113 | |||||||||
| Liabilities associated with assets held for sale | — | — | |||||||||
| Current portion of notes payable | 2,615 | 3,765 | |||||||||
| Total current liabilities | 25,619 | 14,396 | |||||||||
| Operating lease liabilities | 1,827 | 2,845 | |||||||||
| Convertible notes | 187,935 | 221,989 | |||||||||
| Deferred tax liabilities, net | 479 | 479 | |||||||||
| Total long-term liabilities | 190,241 | 225,313 | |||||||||
| Total liabilities | 215,860 | 239,709 | |||||||||
| Equity: | |||||||||||
Common Stock - $0.01 par value, 1,000,000,000 shares authorized; 690,082,283 and 670,585,576 shares issued at December 31, 2021 and 2020, respectively |
6,901 | 6,706 | |||||||||
| Treasury Stock, at cost - 8,655,082 and 549,907 shares at December 31, 2021 and 2020, respectively | (1,791) | (1,791) | |||||||||
| Additional paid-in capital | 3,222,487 | 3,152,694 | |||||||||
| Accumulated other comprehensive income (loss) | (30,495) | (4,225) | |||||||||
| Accumulated deficit | (1,511,976) | (1,481,833) | |||||||||
| Total shareholders’ equity | 1,685,126 | 1,671,551 | |||||||||
| Total liabilities and equity | $ | 1,900,986 | $ | 1,911,260 | |||||||
The accompanying Notes to Parent Company Condensed Financial Statements are an integral part of these statements.
145
OPKO Health, Inc.
PARENT COMPANY CONDENSED STATEMENTS OF INCOME
(In thousands)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Revenues: | |||||||||||||||||
| Revenue from products | $ | — | $ | — | $ | 796 | |||||||||||
| Revenue from transfer of intellectual property and other | — | 1,069 | 1,150 | ||||||||||||||
| Total revenues | — | 1,069 | 1,946 | ||||||||||||||
| Costs and expenses: | |||||||||||||||||
| Costs of revenue | 1,279 | 260 | 1,382 | ||||||||||||||
| Selling, general and administrative | 66,483 | 41,736 | 47,949 | ||||||||||||||
| Research and development | 2,029 | 1,951 | 2,027 | ||||||||||||||
| Total costs and expenses | 69,791 | 43,947 | 51,358 | ||||||||||||||
| Operating loss | (69,791) | (42,878) | (49,412) | ||||||||||||||
| Other income and (expense), net: | |||||||||||||||||
| Interest income | 334 | 967 | 2,186 | ||||||||||||||
| Interest expense | (21,297) | (23,585) | (21,172) | ||||||||||||||
| Fair value changes of derivative instruments, net | (58) | (46) | (601) | ||||||||||||||
| Other income (expense), net | (9,013) | 10,354 | (8,887) | ||||||||||||||
| Other income and (expense), net | (30,034) | (12,310) | (28,474) | ||||||||||||||
| Loss before income taxes and investment losses | (99,825) | (55,188) | (77,886) | ||||||||||||||
| Income tax provision | (2) | (175) | (5) | ||||||||||||||
| Net loss before investment losses | (99,827) | (55,363) | (77,891) | ||||||||||||||
| Loss from investments in investees | (629) | (480) | (2,900) | ||||||||||||||
| Net income (loss) from subsidiaries, net of taxes | 70,313 | 86,429 | (234,134) | ||||||||||||||
| Net income (loss) | $ | (30,143) | $ | 30,586 | $ | (314,925) | |||||||||||
The accompanying Notes to Parent Company Condensed Financial Statements are an integral part of these statements.
146
OPKO Health, Inc.
PARENT COMPANY CONDENSED STATEMENTS OF COMPREHENSIVE INCOME
(In thousands)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Net income (loss) | $ | (30,143) | $ | 30,586 | $ | (314,925) | |||||||||||
| Other comprehensive income (loss), net of tax: | |||||||||||||||||
| Change in foreign currency translation and other comprehensive income (loss) | (26,270) | 17,845 | (1,939) | ||||||||||||||
| Comprehensive income (loss) | $ | (56,413) | $ | 48,431 | $ | (316,864) | |||||||||||
The accompanying Notes to Parent Company Condensed Financial Statements are an integral part of these statements.
147
OPKO Health, Inc.
PARENT COMPANY CONDENSED STATEMENTS OF CASH FLOWS
(In thousands)
| For the years ended December 31, | |||||||||||||||||
| 2021 | 2020 | 2019 | |||||||||||||||
| Cash flows from operating activities: | |||||||||||||||||
| Net income (loss) | $ | (30,143) | $ | 30,586 | $ | (314,925) | |||||||||||
| Adjustments to reconcile net income (loss) to net cash used in operating activities: | |||||||||||||||||
| Depreciation and amortization | — | — | 59 | ||||||||||||||
| Non-cash interest | 9,389 | 9,994 | 8,545 | ||||||||||||||
| Amortization of deferred financing costs | 722 | 787 | 922 | ||||||||||||||
| Losses from investments in investees | 629 | 480 | 2,900 | ||||||||||||||
| (Income) loss from subsidiaries | (70,313) | (86,429) | 234,134 | ||||||||||||||
| Equity-based compensation – employees and non-employees | 13,632 | 8,947 | 13,421 | ||||||||||||||
| Non-cash revenue from the transfer of intellectual property | (3,801) | — | — | ||||||||||||||
| Realized gain on equity securities and disposal of fixed assets | (2,981) | (10,324) | (796) | ||||||||||||||
| Change in fair value of derivative instruments | 4,871 | (6) | 9,523 | ||||||||||||||
| Loss on conversion of the 2025 Notes | 11,111 | — | — | ||||||||||||||
| Adoption of ASC 326 and other | — | (1,311) | — | ||||||||||||||
| Changes in other assets and liabilities | 10,970 | (243) | 3,907 | ||||||||||||||
| Net cash used in operating activities | (55,914) | (47,519) | (42,310) | ||||||||||||||
| Cash flows from investing activities: | |||||||||||||||||
| Investments in investees | (2,000) | — | (1,200) | ||||||||||||||
| Subsidiary financing | 69,608 | (23,234) | (152,376) | ||||||||||||||
| Proceeds from sale of equity securities | 8,078 | 15,110 | — | ||||||||||||||
| Net cash provided by (used in) investing activities | 75,686 | (8,124) | (153,576) | ||||||||||||||
| Cash flows from financing activities: | |||||||||||||||||
| Issuance convertible notes, net | — | — | 200,293 | ||||||||||||||
| Issuance of common stock | — | — | 76,061 | ||||||||||||||
| Debt issuance costs | — | — | (7,762) | ||||||||||||||
| Proceeds from the exercise of Common Stock options and warrants | 1,080 | 756 | (3) | ||||||||||||||
| Borrowings on lines of credit | — | — | 28,800 | ||||||||||||||
| Repayments of lines of credit | — | — | (28,800) | ||||||||||||||
| Redemption of 2033 Senior Notes | — | — | (28,800) | ||||||||||||||
| Net cash provided by financing activities | 1,080 | 756 | 239,789 | ||||||||||||||
| Net increase (decrease) in cash and cash equivalents | 20,852 | (54,887) | 43,903 | ||||||||||||||
| Cash and cash equivalents at beginning of period | 3,740 | 58,627 | 14,724 | ||||||||||||||
| Cash and cash equivalents at end of period | $ | 24,592 | $ | 3,740 | $ | 58,627 | |||||||||||
| SUPPLEMENTAL INFORMATION: | |||||||||||||||||
| Interest paid | $ | 8,515 | $ | 10,908 | $ | 5,224 | |||||||||||
| Income taxes paid, net of refunds | $ | 5,969 | $ | (903) | $ | (1,300) | |||||||||||
| Operating lease right-of-use assets due to adoption of ASU No. 2016-02 | $ | — | $ | — | $ | 4,855 | |||||||||||
| Operating lease liabilities due to adoption of ASU No. 2016-02 | $ | — | $ | — | $ | 4,855 | |||||||||||
| Non-cash financing: | |||||||||||||||||
| Shares issued upon the conversion of: | |||||||||||||||||
| Common Stock options and warrants, surrendered in net exercise | $ | — | $ | — | $ | 20 | |||||||||||
The accompanying Notes to Parent Company Condensed Financial Statements are an integral part of these statements.
148
OPKO Health, Inc.
Notes to Parent Company Condensed Financial Statements
Note 1. Organization and Basis of Presentation
We are a diversified healthcare company that seeks to establish industry-leading positions in large and rapidly growing medical markets. The parent company condensed financial statements included in this Schedule I represent the financial statements of OPKO Health, Inc., the parent company (or “OPKO”), on a stand-alone basis and do not include results of operations from our consolidated subsidiaries. The Parent Company Condensed Financial Statements should be read in conjunction with our audited consolidated financial statements included in Item 8 of Part II of this Form 10-K. As of December 31, 2021 and 2020, approximately $1.9 billion and $1.9 billion, respectively, of our Investments, net have not been eliminated in the parent company condensed financial statements.
The Parent Company Condensed Financial Statements included herein have been prepared in accordance with Rule 12-04, Schedule I of Regulation S-X, as substantially all the assets of BioReference, a wholly-owned subsidiary, and its subsidiaries are restricted from sale, transfer, lease, disposal or distributions to OPKO under the A&R Credit Agreement (as defined below), subject to certain exceptions. BioReference and its subsidiaries’ net assets as of December 31, 2021 were approximately $1.1 billion, which includes goodwill of $283.0 million and intangible assets of $204.4 million. BioReference’s restricted net assets exceeds 25% of OPKO’s consolidated net assets of $1.7 billion as of December 31, 2021.
Note 2 Debt
On February 25, 2020, we entered into a credit agreement with an affiliate of Dr. Frost, pursuant to which the lender committed to provide us with an unsecured line of credit in the amount of $100 million. The line of credit called for a commitment fee equal to 0.25% per annum of the unused portion of the line. We terminated this line of credit in June 2021 and as of December 31, 2021, no amount was outstanding thereunder.
In February 2019, we issued $200.0 million aggregate principal amount of Senior Convertible Notes due 2025 (the “2025 Notes”) in an underwritten public offering. The 2025 Notes bear interest at a rate of 4.50% per year, payable semiannually in arrears on February 15 and August 15 of each year. The 2025 Notes mature on February 15, 2025, unless earlier repurchased, redeemed or converted.
Holders may convert their 2025 Notes into shares of Common Stock at their option at any time prior to the close of business on the business day immediately preceding November 15, 2024 only under the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ended March 31, 2019 (and only during such calendar quarter), if the last reported sale price of our Common Stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; (2) during the five business day period after any five consecutive trading day period (the “measurement period”) in which the trading price per $1,000 principal amount of 2025 Notes for each trading day of the measurement period was less than 98% of the product of the last reported sale price of our Common Stock and the conversion rate on each such trading day; (3) if we call any or all of the 2025 Notes for redemption, at any time prior to the close of business on the scheduled trading day immediately preceding the redemption date; or (4) upon the occurrence of specified corporate events set forth in the indenture governing the 2025 Notes. On or after November 15, 2024, until the close of business on the business day immediately preceding the maturity date, holders of the 2025 Notes may convert their notes at any time, regardless of the foregoing conditions. Upon conversion, we will pay or deliver, as the case may be, cash, shares of our Common Stock, or a combination of cash and shares of our Common Stock, at our election.
The initial and current conversion rate for the 2025 Notes is 236.7424 shares of Common Stock per $1,000 principal amount of 2025 Notes (equivalent to a conversion price of approximately $4.22 per share of Common Stock). The conversion rate for the 2025 Notes is subject to adjustment in certain events, but will not be adjusted for any accrued and unpaid interest. In addition, following certain corporate events that occur prior to the maturity date of the 2025 Notes or if we deliver a notice of redemption, in certain circumstances the indenture governing the 2025 Notes requires an increase in the conversion rate of the 2025 Notes for a holder who elects to convert its notes in connection with such a corporate event or notice of redemption, as the case may be.
We may not redeem the 2025 Notes prior to February 15, 2022. We may redeem for cash any or all of the notes, at our option, on or after February 15, 2022, if the last reported sale price of our Common Stock has been at least 130% of the then current conversion price for the notes for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading day period (including the last trading day of such period) ending on, and including, the trading day immediately
149
preceding the date on which we provide notice of redemption at a redemption price equal to 100% of the principal amount of the notes to be redeemed, plus accrued and unpaid interest to, but excluding, the redemption date. No sinking fund is provided for the 2025 Notes.
If we undergo a fundamental change, as defined in the indenture governing the 2025 Notes, prior to the maturity date of the 2025 Notes, holders may require us to repurchase for cash all or any portion of their notes at a repurchase price equal to 100% of the principal amount of the notes to be repurchased, plus accrued and unpaid interest to, but excluding, the fundamental change repurchase date. The 2025 Notes are our senior unsecured obligations and rank senior in right of payment to any of our indebtedness that is expressly subordinated in right of payment to the 2025 Notes; equal in right of payment to any of our existing and future liabilities that are not so subordinated; effectively junior in right of payment to any of our secured indebtedness to the extent of the value of the assets securing such indebtedness; and structurally junior to all indebtedness and other liabilities (including trade payables) of our current or future subsidiaries.
In May 2021, we entered into exchange agreements with certain holders of the 2025 Notes pursuant to which the holders exchanged $55.4 million in aggregate principal amount of the outstanding 2025 Notes for 19,051,270 shares of our Common Stock (the “Exchange”). We recorded an $11.1 million non-cash loss related to the Exchange.
In conjunction with the issuance of the 2025 Notes, we agreed to loan up to 30,000,000 shares of our Common Stock to affiliates of the underwriter in order to assist investors in the 2025 Notes to hedge their position. Following consummation of the Exchange, the number of outstanding borrowed shares of Common Stock was reduced by approximately 8,105,175 shares. As of December 31, 2021 and 2020, a total of 21,144,825 and 29,250,000 shares were issued under the share lending arrangement, respectively. We will not receive any of the proceeds from the sale of the borrowed shares, but we received a one-time nominal fee of $0.3 million for the newly issued shares. Shares of our Common Stock outstanding under the share lending arrangement are excluded from the calculation of basic and diluted earnings per share. See Note 4.
As required by ASC 470-20, “Debt with Conversion and Other Options,” we calculated the equity component of the 2025 Notes, taking into account both the fair value of the conversion option and the fair value of the share lending arrangement. The equity component was valued at $52.6 million at issue date and this amount was recorded as Additional paid-in capital, which resulted in a discount on the 2025 Notes. The discount is being amortized to Interest expense over the term of the 2025 Notes, which results in an effective interest rate on the 2025 Notes of 11.2%.
The following table sets forth information related to the 2025 Notes which is included in our Consolidated Balance Sheet as of December 31, 2021:
| (In thousands) | 2025 Senior Notes | Discount | Debt Issuance Costs | Total | |||||||||||||||||||
| Balance at December 31, 2020 | $ | 200,000 | $ | (39,537) | $ | (4,300) | $ | 156,163 | |||||||||||||||
| Amortization of debt discount and debt issuance costs | — | 6,639 | 723 | 7,362 | |||||||||||||||||||
| Conversion | (55,420) | 10,151 | 1,104 | (44,165) | |||||||||||||||||||
| Balance at December 31, 2021 | $ | 144,580 | $ | (22,747) | $ | (2,473) | $ | 119,360 | |||||||||||||||
In February 2018, we issued a series of 5% Convertible Promissory Notes (the “2023 Convertible Notes”) in the aggregate principal amount of $55.0 million. The 2023 Convertible Notes mature 5 years from the date of issuance. Each holder of a 2023 Convertible Note has the option, from time to time, to convert all or any portion of the outstanding principal balance of such 2023 Convertible Note, together with accrued and unpaid interest thereon, into shares of our Common Stock at a conversion price of $5.00 per share of Common Stock. We may redeem all or any part of the then issued and outstanding 2023 Convertible Notes, together with accrued and unpaid interest thereon, pro rata among the holders, upon no fewer than 30 days, and no more than 60 days, notice to the holders. The 2023 Convertible Notes contain customary events of default and representations and warranties of OPKO.
Purchasers of the 2023 Convertible Notes included an affiliate of Dr. Phillip Frost, M.D., our Chairman and Chief Executive Officer, and Dr. Jane H. Hsiao, Ph.D., MBA, our Vice-Chairman and Chief Technical Officer.
In January 2013, we entered into note purchase agreements with respect to the issuance and sale of our 3.0% Senior Notes due 2033 (the “2033 Senior Notes”) in a private placement exempt from registration under the Securities Act. We issued the 2033 Senior Notes on January 30, 2013. The 2033 Senior Notes, which totaled $175.0 million in original principal amount, bear interest at the rate of 3.0% per year, payable semiannually on February 1 and August 1 of each year. The 2033 Senior Notes mature on February 1, 2033, unless earlier repurchased, redeemed or converted. Upon a fundamental change as defined in the indenture, governing the 2033 Senior Notes, subject to certain exceptions, the holders may require us to repurchase all or
150
any portion of their 2033 Senior Notes for cash at a repurchase price equal to 100% of the principal amount of the 2033 Senior Notes being repurchased, plus any accrued and unpaid interest to but not including the related fundamental change repurchase date.
From 2013 to 2016, holders of the 2033 Senior Notes converted $143.2 million in aggregate principal amount into an aggregate of 21,539,873 shares of Common Stock. On February 1, 2019, approximately $28.8 million aggregate principal amount of 2033 Senior Notes were tendered by holders pursuant to such holders’ option to require us to repurchase the 2033 Senior Notes as set forth in the indenture, governing the 2033 Senior Notes, following which repurchase only $3.0 million aggregate principal amount of the 2033 Senior Notes remained outstanding. Holders of the remaining $3.0 million principal amount of the 2033 Senior Notes may require us to repurchase such notes for 100% of their principal amount, plus accrued and unpaid interest, on February 1, 2023, on February 1, 2028, or following the occurrence of a fundamental change as described above.
The terms of the 2033 Senior Notes, include, among others: (i) rights to convert the notes into shares of our Common Stock, including upon a fundamental change; and (ii) a coupon make-whole payment in the event of a conversion by the holders of the 2033 Senior Notes on or after February 1, 2017 but prior to February 1, 2019. We determined that these specific terms were embedded derivatives. Embedded derivatives are required to be separated from the host contract, the 2033 Senior Notes, and carried at fair value when: (a) the embedded derivative possesses economic characteristics that are not clearly and closely related to the economic characteristics of the host contract; and (b) a separate, stand-alone instrument with the same terms would qualify as a derivative instrument. We concluded that the embedded derivatives within the 2033 Senior Notes met these criteria and, as such, were valued separate and apart from the 2033 Senior Notes and recorded at fair value each reporting period.
For accounting and financial reporting purposes, we combined these embedded derivatives and valued them together as one unit of accounting. In 2017, certain terms of the embedded derivatives expired pursuant to the original agreement and the embedded derivatives no longer met the criteria to be separated from the host contract and, as a result, the embedded derivatives were no longer required to be valued separate and apart from the 2033 Senior Notes and were reclassified to additional paid in capital.
In November 2015, BioReference and certain of its subsidiaries entered into a credit agreement with JPMorgan Chase Bank, N.A. (“CB”), as lender and administrative agent, as amended (the “Credit Agreement”). The Credit Agreement provides for a $75.0 million secured revolving credit facility and includes a $20.0 million sub-facility for swingline loans and a $20.0 million sub-facility for the issuance of letters of credit.
On August 30, 2021, the Credit Agreement was amended and restated (the “A&R Credit Agreement”). The A&R Credit Agreement is guaranteed by all of BioReference’s domestic subsidiaries. The A&R Credit Agreement is also secured by substantially all assets of BioReference and its domestic subsidiaries, as well as a non-recourse pledge by us of our equity interest in BioReference. Availability under the A&R Credit Agreement is based on a borrowing base composed of eligible accounts receivables of BioReference and certain of its subsidiaries, as specified therein. As of December 31, 2021, $64.8 million remained available for borrowing under the Credit Agreement. Principal under the Credit Agreement is due upon maturity on August 30, 2024.
Note 3 Commitments and Contingencies
See Note 14 of our Consolidated Financial Statements in Item 8 of Part II of this Form 10-K for a discussion of our commitments and contingencies.
Note 4 Dividends
We received $45 million dividend payments from our consolidated subsidiaries for the year ended December 31, 2021. We received a $12 million dividend payment from BioReference during the year ended December 31, 2020.
Note 5 Income Taxes
The Parent Company Condensed Financial Statements recognize the current and deferred income tax consequences that result from our activities during the current and preceding periods pursuant to the provisions of Accounting Standards Codification Topic 740, Income Taxes (ASC 740), as if we were a separate taxpayer rather than a member of the consolidated income tax return group. The tax expense and benefit recorded in OPKO’s consolidated financial statements was the result of activity at the subsidiaries and therefore all tax benefit and expense was reported in the Net income (loss) from subsidiaries, net of taxes line in the Condensed Statement of Income.
Note 6 Subsequent Events
151
We have reviewed all subsequent events and transactions that occurred after the date of our December 31, 2021 Consolidated Balance Sheet date, through the time of filing this Annual Report on Form 10-K.
152
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Date: March 1, 2022 | OPKO HEALTH, INC. | |||||||
| By: | /s/ Phillip Frost, M.D. | |||||||
| Phillip Frost, M.D. | ||||||||
| Chairman of the Board and | ||||||||
| Chief Executive Officer | ||||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||||||||||||
| /s/ Phillip Frost, M.D. | Chairman of the Board and Chief Executive | March 1, 2022 | ||||||||||||
| Phillip Frost, M.D. | Officer (Principal Executive Officer) |
|||||||||||||
| /s/ Jane H. Hsiao, Ph.D., MBA | Vice Chairman and Chief Technical Officer | March 1, 2022 | ||||||||||||
| Jane H. Hsiao, Ph.D., MBA | ||||||||||||||
| /s/ Steven D. Rubin | Director and Executive Vice President – | March 1, 2022 | ||||||||||||
| Steven D. Rubin | Administration | |||||||||||||
| /s/ Adam Logal | Senior Vice President, Chief Financial Officer, | March 1, 2022 | ||||||||||||
| Adam Logal | Chief Accounting Officer and Treasurer (Principal Financial Officer) |
|||||||||||||
| /s/ Jon R. Cohen | Director and Senior Vice President | March 1, 2022 | ||||||||||||
| Jon R. Cohen, M.D. | ||||||||||||||
| /s/ Richard Krasno, Ph.D. | Director | March 1, 2022 | ||||||||||||
| Richard Krasno, Ph.D. | ||||||||||||||
| /s/ Roger J. Medel, M.D. | Director | March 1, 2022 | ||||||||||||
| Roger J. Medel, M.D. | ||||||||||||||
| /s/ John A. Paganelli | Director | March 1, 2022 | ||||||||||||
| John A. Paganelli | ||||||||||||||
| /s/ Richard C. Pfenniger, Jr. | Director | March 1, 2022 | ||||||||||||
| Richard C. Pfenniger, Jr. | ||||||||||||||
| /s/ Alice Lin-Tsing Yu, M.D., Ph.D. | Director | March 1, 2022 | ||||||||||||
| Alice Lin-Tsing Yu, M.D., Ph.D. | ||||||||||||||
153
| Exhibit Number | Description | ||||
| 101.INS | XBRL Instance Document | ||||
| 101.SCH | XBRL Taxonomy Extension Schema Document | ||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | ||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | ||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document | ||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | ||||
154