10-K: Annual report pursuant to Section 13 and 15(d)
Published on March 16, 2011
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number 001-33528
OPKO HEALTH, INC.
(Exact Name of Registrant as Specified in Its Charter)
| DELAWARE | 75-2402409 | |
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) |
4400 Biscayne Blvd., Miami, FL 33137
(Address of Principal Executive Offices, Zip Code)
Registrants Telephone Number, Including Area Code: (305) 575-4100
Securities registered pursuant to section 12(b) of the Act:
| Title of Each Class Common Stock, $.01 par value per share |
Name of Each Exchange on Which Registered NYSE Amex |
Securities registered pursuant to section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule
405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section
13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed
by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or
for such shorter period that the registrant was required to file such reports), and (2) has been
subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark whether the Registrant has submitted electronically and posted on its
corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant
to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the
registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation
S-K is not contained herein, and will not be contained, to the best of registrants knowledge, in
definitive proxy or information statements incorporated by reference in Part III of this Form 10-K
or any amendment to this Form 10-K. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated
filer, a non-accelerated filer, or a smaller reporting company. See the definitions of large
accelerated filer, Accelerated filer and smaller reporting company in Rule 12b-2 of the
Exchange Act.
| Large Accelerated filer o | Accelerated filer þ | Non-Accelerated filer o | Smaller Reporting Company o | |||
| (Do not check if a smaller reporting company) |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of
the Act). Yes o No þ
The aggregate market value of the voting and non-voting common equity held by non-affiliates
computed by reference to the price at which the common equity was last sold, as of the last
business day of the registrants most recently completed second fiscal quarter was: $253,812,039.
As of March 8, 2011 the registrant had 255,600,194 shares of common stock outstanding.
Documents Incorporated by Reference
Portions of the registrants definitive proxy statement for its 2011 Annual Meeting of
Stockholders are incorporated by reference in Items 10, 11, 12, 13, and 14 of Part III of this
Annual Report on Form 10-K.
TABLE OF CONTENTS
| Page | ||||||||
| 6 | ||||||||
| 20 | ||||||||
| 40 | ||||||||
| 40 | ||||||||
| 40 | ||||||||
| 40 | ||||||||
| 41 | ||||||||
| 43 | ||||||||
| 43 | ||||||||
| 50 | ||||||||
| 52 | ||||||||
| 85 | ||||||||
| 85 | ||||||||
| 85 | ||||||||
Item 10. Directors, Executive Officers and Corporate Governance |
||||||||
Item 11. Executive Compensation |
||||||||
Item 12. Security Ownership of Certain Beneficial Owners and Management and
Related Stockholder Matters |
||||||||
Item 13. Certain Relationships and Related Transactions, and Director Independence |
||||||||
Item 14. Principal Accounting Fees and Services |
||||||||
| 87 | ||||||||
| 90 | ||||||||
Certifications |
93 | |||||||
| EX-10.27 | ||||||||
| EX-21 | ||||||||
| EX-23.1 | ||||||||
| EX-31.1 | ||||||||
| EX-31.2 | ||||||||
| EX-32.1 | ||||||||
| EX-32.2 | ||||||||
2
Table of Contents
CAUTIONARY STATEMENTS REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements, as that term is defined
under the Private Securities Litigation Reform Act of 1995 (PSLRA), Section 27A of the Securities
Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended.
Forward-looking statements include statements about our expectations, beliefs or intentions
regarding our product development efforts, business, financial condition, results of operations,
strategies or prospects. You can identify forward-looking statements by the fact that these
statements do not relate strictly to historical or current matters. Rather, forward-looking
statements relate to anticipated or expected events, activities, trends or results as of the date
they are made. Because forward-looking statements relate to matters that have not yet occurred,
these statements are inherently subject to risks and uncertainties that could cause our actual
results to differ materially from any future results expressed or implied by the forward-looking
statements. Many factors could cause our actual activities or results to differ materially from
the activities and results anticipated in forward-looking statements. These factors include those
described below and in Item 1A-Risk Factors of this Annual Report on Form 10-K. We do not
undertake any obligation to update forward-looking statements. We intend that all forward-looking
statements be subject to the safe-harbor provisions of the PSLRA. These forward-looking statements
are only predictions and reflect our views as of the date they are made with respect to future
events and financial performance.
Risks and uncertainties, the occurrence of which could adversely affect our business, include
the following:
| | We have a history of operating losses and we do not expect to become profitable in the near future. | ||
| | Our technologies are in an early stage of development and are unproven. | ||
| | Our drug research and development activities may not result in commercially viable products. | ||
| | The results of previous clinical trials may not be predictive of future results, and our current and planned clinical trials may not satisfy the requirements of the FDA or other non-U.S. regulatory authorities. | ||
| | We will require substantial additional funding, which may not be available to us on acceptable terms, or at all. | ||
| | Our business is substantially dependant on our ability to develop, launch and generate revenue from our molecular diagnostic program. | ||
| | We expect to finance future cash needs primarily through public or private offerings, debt financings or strategic collaborations, which may dilute your stockholdings in the Company. | ||
| | If our competitors develop and market products that are more effective, safer or less expensive than our future product candidates, our commercial opportunities will be negatively impacted. | ||
| | The regulatory approval process is expensive, time consuming and uncertain and may prevent us or our collaboration partners from obtaining approvals for the commercialization of some or all of our product candidates. | ||
| | Failure to recruit and enroll patients for clinical trials may cause the development of our product candidates to be delayed. | ||
| | Even if we obtain regulatory approvals for our product candidates, the terms of approvals and ongoing regulation of our products may limit how we manufacture and market our product candidates, which could materially impair our ability to generate anticipated revenues. | ||
| | We may not meet regulatory quality standards applicable to our manufacturing and quality processes. | ||
| | Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our products. | ||
| | If we fail to attract and retain key management and scientific personnel, we may be unable to successfully develop or commercialize our product candidates. |
3
Table of Contents
| | In the event that we successfully evolve from a company primarily involved in development to a company also involved in commercialization, we may encounter difficulties in managing our growth and expanding our operations successfully. | ||
| | If we fail to acquire and develop other products or product candidates, at all or on commercially reasonable terms, we may be unable to diversify or grow our business. | ||
| | We have no experience manufacturing our pharmaceutical product candidates other than our Mexican facility and we therefore rely on third parties to manufacture and supply our pharmaceutical product candidates, and would need to meet various standards necessary to satisfy FDA regulations if and when we commence manufacturing. | ||
| | We currently have no pharmaceutical or diagnostic marketing, sales or distribution capabilities other than in Chile and Mexico for sales in those countries. If we are unable to develop our sales and marketing and distribution capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our pharmaceutical product candidates. | ||
| | Independent clinical investigators and contract research organizations that we engage to conduct our clinical trials may not be diligent, careful or timely. | ||
| | The success of our business is dependent on the actions of our collaborative partners. | ||
| | Our license agreement with TESARO, Inc. is important to our business. If TESARO does not successfully develop and commercialize rolapitant, our business could be adversely affected. | ||
| | If we are unable to obtain and enforce patent protection for our products, our business could be materially harmed. | ||
| | We do not have an exclusive arrangement in place with Dr. Tom Kodadek with respect to technology or intellectual property that may be material to our business. | ||
| | If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected. | ||
| | We will rely heavily on licenses from third parties. | ||
| | We license patent rights to certain of our technology from third-party owners. If such owners do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects will be harmed. | ||
| | Our commercial success depends significantly on our ability to operate without infringing the patents and other proprietary rights of third parties. | ||
| | Adverse results in material litigation matters or governmental inquiries could have a material adverse effect upon our business and financial condition. | ||
| | Medicare prescription drug coverage legislation and future legislative or regulatory reform of the health care system may affect our ability to sell our products profitably. | ||
| | Failure to obtain regulatory approval outside the United States will prevent us from marketing our product candidates abroad. | ||
| | We may not have the funding available to pursue acquisitions. | ||
| | Acquisitions may disrupt our business, distract our management and may not proceed as planned; and we may encounter difficulties in integrating acquired businesses. | ||
| | Non-U.S. governments often impose strict price controls, which may adversely affect our future profitability. |
4
Table of Contents
| | Our business may become subject to legal, economic, political, regulatory and other risks associated with international operations. | ||
| | The market price of our common stock may fluctuate significantly. | ||
| | Directors, executive officers, principal stockholders and affiliated entities own a significant percentage of our capital stock, and they may make decisions that you do not consider to be in your best interests or in the best interests of our stockholders. | ||
| | Compliance with changing regulations concerning corporate governance and public disclosure may result in additional expenses. | ||
| | If we are unable to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act of 2002, as they apply to us, or our internal controls over financial reporting are not effective, the reliability of our financial statements may be questioned and our common stock price may suffer. | ||
| | We may be unable to maintain our listing on the NYSE Amex, which could cause our stock price to fall and decrease the liquidity of our common stock. | ||
| | Future issuances of common stock and hedging activities may depress the trading price of our common stock. | ||
| | Provisions in our charter documents and Delaware law could discourage an acquisition of us by a third party, even if the acquisition would be favorable to you. | ||
| | We do not intend to pay cash dividends on our common stock in the foreseeable future. |
5
Table of Contents
PART I
Unless the context otherwise requires, all references in this Annual Report on Form 10-K to
the Company, OPKO, we, our, ours, and us refers to OPKO Health, Inc., a Delaware
corporation, including our wholly-owned subsidiaries.
| ITEM 1. | BUSINESS |
OVERVIEW
We are a multi-national pharmaceutical and diagnostics company that seeks to establish
industry-leading positions in large and rapidly growing medical markets by leveraging our
discovery, development and commercialization expertise and our novel and proprietary technologies.
Our current focus is on conditions with major unmet medical needs including neurological disorders,
infectious diseases, oncology and ophthalmologic diseases. We are developing a range of solutions
to diagnose, treat and prevent these conditions, including molecular diagnostics tests, proprietary
pharmaceuticals and vaccines. We plan to commercialize these solutions on a global basis in large
and high growth markets, including emerging markets. We have already established emerging markets
pharmaceutical platforms in Chile and Mexico, which are delivering revenue and which we expect to
deliver cash flow and facilitate future market entry for our products currently in development. We
also actively explore opportunities to acquire complementary pharmaceuticals, compounds,
technologies, and businesses.
Our lead program under development is an innovative molecular diagnostic platform technology
for the rapid identification of molecules or immunobiomarkers that may be useful in the creation of
accurate, easy-to-use tests for conditions where we believe no objective diagnostic test currently
exists or where presently available tests are characterized by invasive procedures and low levels
of accuracy. We have demonstrated in initial studies that our platform has the ability to identify
diagnostic biomarkers for a wide range of diseases to which the immune system reacts, including
cancers, autoimmune diseases, neurodegenerative diseases and infectious diseases. This technology
platform may also allow for the development of vaccines and highly targeted therapeutic agents.
Our most advanced application of this technology is a simple blood test for Alzheimers
disease, a debilitating neurodegenerative disease for which there are limited diagnostic options
available today. Based on initial clinical work, as described in the journal Cell in January 2011,
our Alzheimers test demonstrated an ability to identify and differentiate Alzheimers patients by
detecting elevated levels of antibodies that appear to be unique to Alzheimers disease. We are
currently conducting a broader validation study that we expect to be completed by late 2011 and we
expect to begin marketing our test for Alzheimers disease in 2013. We believe that this test
could initially be useful in stratifying patients for ongoing clinical trials of potential
Alzheimers drugs as well as to confirm the diagnosis in a clinical setting and to track the
progression of the disease or effectiveness of a therapeutic in a clinical trial. In December 2010
we entered into a non-exclusive collaboration agreement with
Bristol-Myers Squibb Company (BMS)
to investigate the utility of our diagnostic technology for the diagnosis of Alzheimers disease
and for identifying individuals with early stage cognitive impairment that are likely to progress
to Alzheimers disease.
In addition to Alzheimers disease, we are developing a pipeline of diagnostic tests for other
conditions such as pancreatic cancer, Parkinsons disease and non-small cell lung cancer. We
anticipate entering into additional collaboration agreements regarding our diagnostic pipeline
tests and expect to commercially launch up to three diagnostic tests over the next three years.
Our product pipeline also includes several pharmaceutical compounds and technologies in
research and development for a broad range of indications and conditions. We are developing a
protein-based influenza vaccine designed to offer multi-season and multi-strain protection, that we
believe will offer more effective and longer lasting protection against influenza, in addition to
more rapid and efficient production than existing influenza vaccine technologies. We recently
acquired an up-regulating oligonucleotide therapeutics technology that has the potential to create
new drugs for the treatment of a wide variety of illnesses, including cancer, heart disease,
metabolic disorders and a range of genetic disorders. We have a variety of therapeutic agents for
respiratory disorders in clinical development, including products for asthma, chronic obstructive
pulmonary disease (COPD), and chronic cough. In addition to these development programs, we have
growing pharmaceutical businesses in Chile and Mexico.
We have a highly experienced management team that we believe has demonstrated an ability to
successfully build and manage pharmaceutical businesses. Our Chairman and Chief Executive Officer,
Dr. Phillip Frost, founded
6
Table of Contents
and served as Chairman and Chief Executive Officer of IVAX Corporation (IVAX), a
multi-national pharmaceutical company, from 1987 until the acquisition of IVAX by Teva
Pharmaceutical Industries, Limited (Teva), in January 2006. Dr. Frost currently serves as
Chairman of the Board of Teva. Prior to Ivax, Dr. Frost founded and served as Chairman of the
Board of Directors of Key Pharmaceuticals, Inc. from 1972 until the acquisition of Key
Pharmaceuticals by Schering Plough Corporation in 1986. Our other senior executive officers,
including Dr. Jane Hsiao, our Vice Chairman and Chief Technology Officer, Steven Rubin, our
Executive Vice President, Administration, and Dr. Rao Uppaluri, our Senior Vice President and Chief
Financial Officer, are former executive officers of IVAX. Based on their experience in the
industry, we believe that our management team has extensive development, regulatory and
commercialization expertise and relationships that provide access to commercial opportunities.
GROWTH STRATEGY
We expect our future growth to come from leveraging our proprietary technology and development
strengths, and opportunistically pursuing complementary, accretive, or strategic acquisitions and
investments.
We have under development a broad and diversified portfolio of diagnostic tests, vaccines and
small molecules, targeting a broad range of unmet medical needs. We intend to continue to leverage
our proprietary technology and our strengths in all phases of pharmaceutical research and
development to further develop and commercialize our portfolio of proprietary pharmaceutical and
diagnostic products. Key elements of our strategy are to:
| | obtain requisite regulatory approval and compile clinical data for our most advanced product candidates; | ||
| | develop a focused commercialization capability in the United States; | ||
| | strategically utilize our research and development resources to advance our product pipeline; and | ||
| | expand into other medical markets which provide significant opportunities and which we believe are complementary to and synergistic with our business. |
We have and expect to continue to be opportunistic and pursue complementary, or strategic
acquisitions, licenses and investments. Our management team has significant experience in
identifying, executing and integrating these transactions. We expect to use well-timed, carefully
selected acquisitions, licenses and investments to continue to drive our growth, including:
| | Products and technologies. We intend to pursue product and technology acquisitions and licenses that will complement our existing businesses and provide new product and market opportunities, improve our growth, enhance our profitability, leverage our existing assets, and contribute to our own organic growth. | ||
| | Commercial businesses. We intend to continue to pursue acquisitions of commercial businesses that will both drive our growth and provide geographically diverse sales and distribution opportunities, particularly outside of the United States. | ||
| | Early stage investments. We have and may continue to make investments in early stage companies that we perceive to have valuable proprietary technology and significant potential to create value for OPKO as a shareholder. |
Corporate Information
We were originally incorporated in Delaware in October 1991 under the name Cytoclonal
Pharmaceutics, Inc., which was later changed to eXegenics, Inc. On March 27, 2007 we were part of
a three-way merger with Froptix Corporation (Froptix), a research and development company, and
Acuity Pharmaceuticals, Inc. (Acuity), a research and development company. This transaction was
accounted for as a reverse merger between Froptix and eXegenics, with the combined company then
acquiring Acuity. eXegenics was previously involved in the research, creation, and development of
drugs for the treatment and prevention of cancer and infectious diseases; however, eXegenics had
been a public shell company without any operations since 2003. On June 8, 2007 we changed our name
to OPKO Health, Inc.
Our shares are publicly traded on the NYSE Amex under the ticker OPK. Our principal
executive offices are located in Miami, Florida. We also have leased lab space at The Scripps
Research Institute in Jupiter, Florida, and
7
Table of Contents
leased offices in Santiago, Chile. We also have offices and a manufacturing facility in
Guadalajara, Mexico, a leased manufacturing facility in Hialeah, Florida, and a research and
development office in the United Kingdom at the University of Kent.
We currently manage our operations in two reportable segments, pharmaceutical and
instrumentation segments. The pharmaceutical segment consists of two operating segments, (i) our
pharmaceutical research and development segment which is focused on the research and development of
pharmaceutical products, diagnostic tests, and vaccines, and (ii) the pharmaceutical operations we
acquired in Chile and Mexico through the acquisition of OPKO Chile and Exakta-OPKO. The
instrumentation segment consists of ophthalmic instrumentation devices and the activities related
to the research, development, manufacture, and commercialization of those products.
CURRENT PRODUCT CANDIDATES AND RELATED MARKETS
Molecular Diagnostics
In June 2009, we acquired exclusive, worldwide rights from the University of Texas
Southwestern to an innovative platform technology for the rapid identification of molecules or
immunobiomarkers that may be useful in the creation of accurate, easy-to-use diagnostic tests as
well as the development of vaccines and highly targeted therapeutic agents for immune system-driven
diseases. The technology is based on an innovative method for the identification in small blood
samples of disease-specific antibodies that can serve as diagnostic biomarkers for various
diseases. We jointly own patent applications covering certain aspects of the technology and hold
an exclusive license to the technology.
We believe this innovative technology could have broad applicability for the development of
simple and accurate, quantitative blood tests across numerous important diseases, including a
number of disease segments where there are no widely accepted or effective screening tests
available. The first diagnostic product we are pursuing utilizing this technology is a simple
blood test for Alzheimers disease. The test is designed to detect elevated levels of antibodies
that appear to be unique to Alzheimers disease and could be useful in stratifying patients for
ongoing clinical trials of potential Alzheimers drugs as well as to confirm the diagnosis in a
clinical setting and to track the progression of the disease or effectiveness of a therapeutic in a
clinical trial. The Alzheimers disease-specific antibodies were discovered using this novel
proprietary platform that we have demonstrated in initial studies to be capable of identifying
biomarkers for a wide range of diseases to which the immune system reacts, including Alzheimers
disease, as well as cancers, autoimmune diseases, neurodegenerative diseases and infectious
diseases.
Currently it is estimated that over five million people in the United States, and over 35
million people worldwide, have Alzheimers disease and the national cost of caring for people with
Alzheimers and other dementias is estimated to be $172 billion in 2010 in the United States alone.
By 2050, it is estimated that between 11 and 16 million people in the United States over the age
of 65 will have Alzheimers, and the global prevalence of people living with Alzheimers and other
dementias is expected to be greater than 115 million. Currently there are no specific tests to
detect Alzheimers disease and follow its progression. Current diagnosis tools such as behavioral
and cognitive measurements, brain scans and spinal fluid analysis have limited diagnostic accuracy,
may not detect early stage disease, and in the case of spinal fluid analysis are highly invasive.
Definitive diagnosis can currently be made only from examination of postmortem brain tissue
samples. An effective early diagnostic blood test would provide a significant breakthrough in
supporting definitive early diagnosis.
As reported in the January 2011 edition of the journal Cell, we demonstrated in a preliminary
study that we were able to identify unique biomarkers from serum samples of known Alzheimers
disease patients, and then using these biomarkers we were able to distinguish patients with
Alzheimers disease from healthy controls, patients with Parkinsons disease and patients with
lupus. In December 2010, we entered into a collaboration agreement with BMS, under which we and
BMS will investigate the utility of our novel technology for the diagnosis of Alzheimers disease
and for identifying individuals with early stage cognitive impairment that are likely to progress
to Alzheimers disease. We have conducted a validation study of 140 patients, and we are expanding
the study to include 200 patients with Alzheimers disease, 200 demographically matched controls,
and 180 patients with other conditions. We expect to complete this study by late 2011 and we
expect to begin marketing our diagnostic test for Alzheimers disease in 2013.
In addition to Alzheimers disease, we are also pursuing the development of diagnostic tests
for pancreatic cancer, Parkinsons disease, non-small cell lung cancer, and other diseases for
which early detection could lead to earlier therapy and dramatically improved outcomes. We have
conducted preliminary studies in pancreatic cancer,
8
Table of Contents
Parkinsons disease, and non-small cell lung cancer patient samples that we believe
demonstrate the ability of our technology to identify biomarkers with diagnostic utility for these
conditions. We plan to conduct additional studies in larger patient populations to further
validate diagnostic tests for these and other conditions. We expect to complete a validation study
of an initial cancer diagnostic test in 2012 and we expect to begin marketing an initial cancer
diagnostic test in 2013. We anticipate entering into additional collaboration agreements regarding
our diagnostic pipeline tests and expect to commercially launch up to three diagnostic tests over
the next three years.
Along with molecular diagnostic applications, we believe that this same platform technology
should permit the development of pharmaceutical agents or other therapeutics which can be delivered
directly to the targeted autoimmune cells. Similarly, we believe that the synthetic molecules that
we are able to identify through this technology could be used for the formulation of synthetic
vaccines to induce an immune response that protects against foreign pathogens.
Pharmaceutical Business
We presently have several pharmaceutical compounds and technologies in research and
development for a broad range of indications and conditions. Our product development candidates
are in various stages of development. Our primary focus is on developing and commercializing our
novel influenza vaccine and therapeutics based on our oligonucleotide technology platform.
Vaccine Programs
In July 2009, we acquired worldwide rights from Academia Sinica in Taipei, Taiwan, for a new
technology to develop protein-based vaccines against influenza and other viral infections. We are
developing a proprietary, innovative influenza vaccine designed to provide multi-season and
multi-strain protection against many human influenza virus strains, including both seasonal
influenza strains as well as global influenza pandemic strains, such as swine flu (H1N1), and
avian flu (H5N1). The world-wide seasonal influenza market place is projected to increase to
$6.3 billion by 2014. Influenza results in approximately 200,000 hospitalizations and more than
36,000 deaths each year in the United States alone, with estimated economic costs in excess of $87
billion per year.
There are several major limitations of current influenza vaccines, including:
| | Inability to respond to mutations. The influenza virus undergoes frequent and unpredictable antigenic changes, or mutations, in its surface proteins, creating new strains of the virus which the immune system often fails to recognize. Currently available vaccines do not provide adequate protection against new influenza strains, leading to the need for the ongoing development and administration of vaccines on an annual basis. | ||
| | Slow development timelines. Currently available influenza vaccines are based on annual World Health Organization predictions of the influenza strains that will be prevalent in the upcoming season. Because of the long development timeline required to create current influenza vaccines, the actual virus strains prevalent in a given season may differ from the strains used to create the vaccine, resulting in commercially available vaccines that offer limited protection and clinical efficacy. | ||
| | Production cycle limitations. The annual strain prediction and selection process necessitates annual vaccine manufacturing with time-consuming and expensive annual production cycles. The prediction of optimal production quantities is also difficult and often results in either a shortage or excess of doses. |
Instead of the typical method of making a cocktail of inactivated viruses for annual flu
shots, our approach to anti-viral vaccines is designed to increase protective antibodies against
multiple strains of viral influenza. We believe that our technology will, among other things,
permit the development of a molecular protein-based flu vaccine that will provide protection
against multiple H1, H3 or H5 flu variances. We believe that our novel vaccine technology
addresses the current limitations by providing a wider scope of virus strain coverage with
longer-term protection, in a recombinant protein format that requires shorter development timelines
and enables efficient year-round and demand-based production.
In addition, in March 2010, we acquired worldwide rights from Academia Sinica to certain
alpha-galactosyl ceramide analogs which are believed to be useful as vaccines or vaccine adjuvants
for a wide variety of disorders including cancer, infectious disease, and autoimmune disease. We
are working in conjunction with Academia Sinica to advance and develop products under these
technologies.
9
Table of Contents
Oligonucleotide Therapeutics
In January 2011, we acquired CURNA, Inc., a privately held company based in Jupiter, Florida,
engaged in the discovery of new drugs for the treatment of a wide variety of illnesses, including
cancer, heart disease, metabolic disorders and a range of genetic anomalies. CURNAs broad
platform technology utilizes a short, single strand oligonucleotide and is based on the
up-regulation of protein production through interference with non-coding RNAs, or natural
antisense. This strategy contrasts with established approaches which down-regulate protein
production. CURNA has designed a novel type of therapeutic modality, termed AntagoNAT, and has
initially demonstrated this approach for up-regulation of several therapeutically relevant proteins
in in vitro and animal models. We believe that this short, single strand oligonucleotide can be
delivered intravenously or subcutaneously without the drug delivery or cell penetration
complications typically associated with double stranded siRNA therapeutics. CURNA has identified
and developed compounds which increase the production of over 80 key proteins involved in a large
number of individual diseases.
Asthma and COPD
In May 2010, we acquired worldwide rights to a novel heparin-derived oligosaccharide which has
significant potential in treating asthma and COPD. Over 22 million people in the United States
live with asthma, including nearly six million children. Additionally, there are more than 12
million people in the United States who have COPD. The market for asthma and COPD treatments was
estimated to be $26 billion in 2009. Currently available therapies often include unwanted side
effects and may have limited efficacy. We believe that our product may have an improved efficacy
and side effect profile. Our initial studies have demonstrated anti-inflammatory and anti-allergic
activity when administered orally or inhaled with inhalers or nebulizers in sheep and mice asthma
models. We have also successfully completed human feasibility studies in asthma.
NK-1 Program
In November 2009, we acquired rolapitant and other neurokinin-1 (NK-1), assets from Schering
Plough Corporation. Rolapitant, a potent and selective competitive antagonist of the NK-1
receptor, has successfully completed Phase II clinical testing for prevention of chemotherapy
induced nausea and vomiting (CINV), and post-operative induced nausea and vomiting (PONV).
Based on studies conducted to-date, we believe that rolapitant may be differentiated from other
agents in this class through both its duration of action and lack of drug-drug interactions.
Rolapitant has an extended plasma half-life that has the potential to improve the management of
nausea and vomiting experienced by cancer patients undergoing chemotherapy treatment. Phase II
clinical testing of rolapitant for the prevention of nausea and vomiting in cancer patients treated
with highly emetogenic chemotherapy demonstrated promising five-day activity following the
administration of a single dose, with no significant drug-drug interactions.
The global emesis market was nearly $2.4 billion in 2009. There are more than two million
chemotherapy patients each year in the United States, Europe, and Japan alone, and there are more
than 23 million surgery patients in the United States and Europe. NK-1 receptor antagonists and
5HT3 receptor antagonists are major classes of drugs used for prevention of nausea and vomiting.
In general, NK-1 inhibitors are complementary to 5HT3 inhibitors with the potential for additive
effects in PONV and demonstrated additive effects in CINV. While there are several approved 5HT3
receptor antagonists, including palonosetron (Aloxi), ondansetron (Zofran), and other generics,
there is only one NK-1 receptor antagonist approved for commercial use, aprepitant (Emend).
In December 2010, we exclusively out-licensed the development, manufacture and
commercialization of rolapitant to TESARO, Inc., an oncology-focused biopharmaceutical company
co-founded by former executives of MGI PHARMA, an oncology and acute-care focused biopharmaceutical
company acquired by Eisai Co., Ltd. in 2008. We believe that the TESARO team brings significant
development and commercialization experience and a demonstrated track record of success in
launching and differentiating products for the CINV market.
TESARO is initially pursuing development and commercialization of rolapitant for CINV. Under
the terms of the license, we are eligible to receive payments of up to $121.0 million, of which an
up-front payment of $6.0 million has been received, and additional payments based upon net sales
and achievement of specified regulatory and commercialization milestones. In addition, TESARO will
pay us double digit tiered royalties on sales of licensed product. Further, we will share with
TESARO future profits from the commercialization of licensed products in Japan, and we will have an
option to market the products in Latin America. In addition, we acquired an approximately 10%
equity position in TESARO on an as-converted basis.
10
Table of Contents
Separately, we are also developing a second generation NK-1 receptor antagonist, SCH 900978,
for chronic cough. The product has completed a Phase II proof of concept study with no safety
issues identified and low drug-drug interaction potential.
Ophthalmics
We have therapeutic programs under development for a range of ophthalmic diseases and
conditions such as wet and dry Age Related Macular Degeneration (AMD), which represent markets
with significant unmet needs. In July 2007, we initiated the first of two required pivotal Phase
III trials for our lead ophthalmic product, bevasiranib, a drug candidate in development for the
treatment of Wet AMD. On March 6, 2009, following the recommendation of an independent data
monitoring committee (IDMC), we determined to terminate the Phase III clinical trial of
bevasiranib. Review of the data by the IDMC had indicated that the trial as structured was
unlikely to meet its primary end point. We are continuing to investigate improved drug delivery
methods in an effort to determine appropriate next steps regarding the development of bevasiranib.
We may seek to continue development of these programs in the future, or to outlicense or sell these
programs.
Emerging Markets Operations
We also intend to continue to leverage our global commercialization expertise to pursue
acquisitions of commercial businesses that will both drive our growth and provide geographically
diverse sales and distribution opportunities, particularly outside of the United States. It is
estimated that by 2030 emerging markets will account for 60% of global GDP. According to IMS
Health, emerging healthcare markets, including markets such as Brazil, Chile, China, India, Mexico,
Russia, and Turkey, are projected to grow approximately 15% in total per year through 2014, while
developed markets are projected to grow only 3% to 5% over the same period. At a time of slowing
pharmaceutical sales growth in many mature countries, this expansion in many emerging markets has
led to higher sales growth rates and an increasing contribution to the industrys global
performance. As a result we expect that emerging markets will continue to be a growing part of our
business strategy, contributing both attractive revenue growth and cash flow to support our
development programs.
In February 2010, we completed the acquisition of Pharmacos Exakta S.A. de C.V.
(Exakta-OPKO), a Mexican pharmaceutical business engaged in the manufacture, marketing, sale, and
distribution of ophthalmic and other pharmaceutical products to private and public customers in
Mexico. Exakta-OPKO manufacturers and sells more than 25 products primarily in the generics market
in Mexico, although it has recently increased its focus on the development of proprietary products
as well. Exakta-OPKO has also signed a letter of intent to collaborate with the Centro de
Investigación y Asistencia Tecnológica y Diseño del Estado de Jalisco (CIATEJ), a preeminent
technology and research center in the State of Jalisco, Mexico to develop and manufacture vaccines
for flu, dengue fever, and West Nile virus. The first project under development with CIATEJ is a
new H1N1 vaccine which is expected to launch in Mexico in 2012.
In October 2009, we completed the acquisition of Pharma Genexx, S.A. (OPKO Chile). OPKO
Chile markets, sells and distributes more than 100 products in the generics market to
private, hospital and institutional clients in Chile for a wide range of indications, including,
cardiovascular products, vaccines, antibiotics, gastro-intestinal products, and hormones, among
others. OPKO Chile has no manufacturing facility.
Strategic Investments
We have and may continue to make investments in other early stage companies that we perceive
to have valuable proprietary technology and significant potential to create value for OPKO as a
shareholder.
| | In December 2010, we acquired a minority equity interest in TESARO, Inc., a privately held oncology-focused biopharmaceutical company, as part of a license agreement with TESARO for the development, manufacture, commercialization and distribution of rolapitant and a related compound. As of December 31, 2010, we owned an approximately 10% equity position in TESARO on an as-converted basis. | ||
| | In November 2010, we acquired a minority equity interest in Fabrus, LLC, a privately held early-stage biotechnology company with next generation therapeutic antibody drug discovery and development capabilities that is using its proprietary antibody screening and engineering approach to discover promising lead compounds against several important oncology targets. As of December 31, 2010, we owned approximately 13% of the outstanding membership interests of Fabrus. |
11
Table of Contents
| | In September 2009, we acquired a minority equity interest in Cocrystal Discovery, Inc., a privately held biopharmaceutical company focused on the discovery and development of novel small molecule antiviral therapeutics tailored for the treatment of serious and chronic viral diseases. As of December 31, 2010, we owned approximately 16% of the outstanding capital stock of Cocrystal Discovery. | ||
| | In June 2009 we acquired a minority equity interest in Sorrento Therapeutics, Inc., a publicly held development-stage biopharmaceutical company focused on applying its proprietary technology platform for the discovery and development of human therapeutic antibodies for the treatment of a variety of disease conditions, including cancer, inflammation, metabolic disease and infectious disease. As of December 31, 2010 we owned approximately 21% of the outstanding capital stock of Sorrento Therapeutics. |
INSTRUMENTATION BUSINESS
Our instrumentation business consists of the development, commercialization and sale of
ophthalmic diagnostic and imaging systems and instrumentation products. Currently, the
instrumentation business is primarily based on technology that offers innovative systems with
advanced diagnostic imaging capabilities and tools to meet the needs of eye care professionals. We
may seek to continue development of this business in the future or to outlicense or sell this
business.
RESEARCH AND DEVELOPMENT EXPENSES
During the years ended December 31, 2010, 2009, and 2008, we incurred $7.9 million, $12.9
million, and $21.6 million, respectively, of research and development expenses related to our
various product candidates. During the year ended December 31, 2010, our research and development
expense consisted of activities related to the development of our molecular diagnostics program,
rolapitant prior to its divesture, and our next generation OCT/SLO. Research and development
expense for the years ended December 31, 2009 and 2008 primarily relate to bevasiranib. In
addition, during 2009 and 2008, we expensed $2.0 million and $1.4 million for acquired in process
research and development related to our acquisitions of the NK-1 compounds and Vidus Ocular, Inc.
INTELLECTUAL PROPERTY
We believe that technology innovation is driving breakthroughs in healthcare. We have adopted
a comprehensive intellectual property strategy which blends the efforts to innovate in a focused
manner with the efforts of our business development activities to strategically in-license
intellectual property rights. We develop, protect, and defend our own intellectual property rights
as dictated by the developing competitive environment. We value our intellectual property assets
and believe we have benefited from early and insightful efforts at understanding the disease and
the molecular basis of potential pharmaceutical intervention.
We actively seek, when appropriate and available, protection for our products and proprietary
information by means of United States and foreign patents, trademarks, trade secrets, copyrights,
and contractual arrangements. Patent protection in the pharmaceutical field, however, can involve
complex legal and factual issues. There can be no assurance that any steps taken to protect such
proprietary information will be effective.
Because the patent positions of pharmaceutical and biotechnology companies are highly
uncertain and involve complex legal and factual questions, the patents owned and licensed by us, or
any future patents, may not prevent other companies from developing similar or therapeutically
equivalent products or ensure that others will not be issued patents that may prevent the sale of
our products or require licensing and the payment of significant fees or royalties. Furthermore,
to the extent that any of our future products or methods are not patentable, that such products or
methods infringe upon the patents of third parties, or that our patents or future patents fail to
give us an exclusive position in the subject matter claimed by those patents, we will be adversely
affected. We may be unable to avoid infringement of third party patents and may have to obtain a
license, defend an infringement action, or challenge the validity of the patents in court. A
license may be unavailable on terms and conditions acceptable to us, if at all. Patent litigation
is costly and time consuming, and we may be unable to prevail in any such patent litigation or
devote sufficient resources to even pursue such litigation.
LICENSES AND COLLABORATIVE RELATIONSHIPS
Our strategy is to develop a portfolio of product candidates through a combination of internal
development, acquisition, and external partnerships. Collaborations are key to our strategy and we
continue to build relationships
12
Table of Contents
and forge partnerships in various areas where unmet medical need
and commercial opportunities exist. In 2010, we
completed a strategic licensing transaction pursuant to which we exclusively out-licensed
development, manufacture and commercialization of rolapitant to TESARO, an oncology-focused
biopharmaceutical company founded by executives with a demonstrated track record in launching
successful products for the CINV market. Previously, we also completed strategic licensing
transactions with the University of Texas Southwestern Medical Center at Dallas. Academia Sinica,
the Trustees of the University of Pennsylvania, and the University of Florida Research Foundation,
among others.
COMPETITION
The pharmaceutical, molecular diagnostic, and instrumentation industries are highly
competitive and require an ongoing, extensive search for technological innovation. The industries
are characterized by rapidly advancing technologies, intense competition and a strong emphasis on
proprietary products. They also require, among other things, the ability to effectively discover,
develop, test and obtain regulatory approvals for products, as well as the ability to effectively
commercialize, market and promote approved products.
We intend to leverage our technological innovation and proprietary position to effectively
compete in the pharmaceutical and biopharmaceutical markets. In addition, we are committed to
researching, developing and pursuing the commercialization of diagnostic tests for Alzheimers
disease, various cancers and autoimmune disease, among others. Numerous companies, however,
including major pharmaceutical companies, specialty pharmaceutical companies and specialized
biotechnology companies, are engaged in the development, manufacture and marketing of
pharmaceutical products competitive with those that we intend to commercialize ourselves and
through our partners, For example, Merck currently markets Emend, an NK-1 compound for
post-operative nausea and vomiting and chemotherapy induced nausea and vomiting. There are several
companies working to develop universal flu vaccines, and several companies have products or
development programs for diseases and conditions our ophthalmic product candidates are designed to
address. Competitors to our molecular diagnostics business are many and include major diagnostic
companies, reference laboratories, molecular diagnostic firms, universities and research
institutions.
Most of these companies have substantially greater financial and other resources, larger
research and development staffs and more extensive marketing and manufacturing organizations than
ours. This enables them, among other things, to make greater research and development investments
and efficiently utilize their research and development costs, as well as their marketing and
promotion costs, over a broader revenue base. This also provides our competitors with a
competitive advantage in connection with the highly competitive product acquisition and product
in-licensing process, which may include auctions in which the highest bidder wins. Our competitors
may also have more experience and expertise in obtaining marketing approvals from the Food and Drug
Administration (the FDA) and other regulatory authorities. In addition to product development,
testing, approval, and promotion, other competitive factors in the pharmaceutical industry include
industry consolidation, product quality and price, product technology, reputation, customer
service, and access to technical information.
Our ability to commercialize our pharmaceutical and diagnostic test product candidates and
compete effectively will depend, in large part, on:
| | Our ability to meet all necessary regulatory requirements to advance our product candidates through clinical trials and the FDA approval process; | ||
| | the perception by physicians and other members of the health care community of the safety, efficacy, and benefits of our products compared to those of competing products or therapies; | ||
| | our ability to manufacture products we may develop on a commercial scale; | ||
| | the effectiveness of our sales and marketing efforts; | ||
| | the willingness of physicians to adopt a new treatment regimen represented by our technology; | ||
| | our ability to secure reimbursement for our product candidates, | ||
| | the price of the products we may develop and commercialize relative to competing products; |
13
Table of Contents
| | our ability to accurately forecast and meet demand for our product candidates if regulatory approvals are achieved; | ||
| | our ability to develop a commercial scale infrastructure either on our own or with a collaborator, which would include expansion of existing facilities, including our manufacturing facilities, development of a distribution network, and other operational and financial systems necessary to support our increased scale; | ||
| | our ability to maintain a proprietary position in our technologies; and | ||
| | our ability to rapidly expand the existing information technology infrastructure and configure existing operational, manufacturing, and financial systems (on our own or with third party collaborators) necessary to support our increased scale, which would include existing or additional facilities and or partners. |
GOVERNMENT REGULATION OF OUR DRUG AND DEVICE DEVELOPMENT ACTIVITIES
The U.S. government regulates healthcare through various agencies, including but not limited
to the following: (i) the U.S. Food and Drug Administration (FDA), which administers the Federal
Food, Drug and Cosmetic Act (FDCA), as well as other relevant laws; (ii) the Centers for Medicare
& Medicaid Services (CMS), which administers the Medicare and Medicaid programs; (iii) the Office
of Inspector General (OIG), which enforces various laws aimed at curtailing fraudulent or abusive
practices, including by way of example, the Anti-Kickback Law, the Physician Self-Referral Law,
commonly referred to as the Stark law, the Anti-Inducement Law, the Civil Money Penalty Law, and
the laws that authorize the OIG to exclude healthcare providers and others from participating in
federal healthcare programs; and (iv) the Office of Civil Rights, which administers the privacy
aspects of the Health Insurance Portability and Accountability Act of 1996 (HIPAA). All of the
aforementioned are agencies within the Department of Health and Human Services (HHS). Healthcare
is also provided or regulated, as the case may be, by the Department of Defense through its TriCare
program, the Department of Veterans Affairs, especially through the Veterans Health Care Act of
1992, the Public Health Service within HHS under Public Health Service Act § 340B (42 U.S.C. §
256b), the Department of Justice through the Federal False Claims Act and various criminal
statutes, and state governments under the Medicaid and other state sponsored or funded programs and
their internal laws regulating all healthcare activities.
The testing, manufacture, distribution, advertising, and marketing of drug products and
medical devices are subject to extensive regulation by federal, state, and local governmental
authorities in the United States, including the FDA, and by similar agencies in other countries.
Any drug or device product that we develop must receive all relevant regulatory approvals or
clearances, as the case may be, before it may be marketed in a particular country.
Drug Development
The regulatory process, which includes overseeing preclinical studies and clinical trials of
each pharmaceutical compound to establish its safety and efficacy and confirmation by the FDA that
good laboratory, clinical, and manufacturing practices were maintained during testing and
manufacturing, can take many years, requires the expenditure of substantial resources, and gives
larger companies with greater financial resources a competitive advantage over us. Delays or
terminations of clinical trials that we undertake would likely impair our development of product
candidates. Delays or terminations could result from a number of factors, including stringent
enrollment criteria, slow rate of enrollment, size of patient population, having to compete with
other clinical trials for eligible patients, geographical considerations, and others.
The FDA review processes can be lengthy and unpredictable, and we may encounter delays or
rejections of our applications when submitted. Generally, in order to gain FDA approval, we must
first conduct preclinical studies in a laboratory and in animal models to obtain preliminary
information on a compound and to identify any safety problems. The results of these studies are
submitted as part of an IND application that the FDA must review before human clinical trials of an
investigational drug can commence.
Clinical trials are normally done in three sequential phases and generally take two to five
years or longer to complete. Phase I consists of testing the drug product in a small number of
humans, normally healthy volunteers, to determine preliminary safety and tolerable dose range.
Phase II usually involves studies in a limited patient population to evaluate the effectiveness of
the drug product in humans having the disease or medical condition for which the product is
indicated, determine dosage tolerance and optimal dosage, and identify possible common
14
Table of Contents
adverse
effects and safety risks. Phase III consists of additional controlled testing at multiple clinical
sites to establish clinical safety and effectiveness in an expanded patient population of
geographically dispersed test sites to evaluate the overall benefit-risk relationship for
administering the product and to provide an adequate basis for
product labeling. Phase IV clinical trials may be conducted after approval to gain additional
experience from the treatment of patients in the intended therapeutic indication.
After completion of clinical trials of a new drug product, FDA and foreign regulatory
authority marketing approval must be obtained. Assuming that the clinical data support the
products safety and effectiveness for its intended use, a new drug application (NDA), is
submitted to the FDA for its review. Generally, it takes one to three years to obtain approval.
If questions arise during the FDA review process, approval may take a significantly longer period
of time. The testing and approval processes require substantial time and effort and we may not
receive approval on a timely basis, if at all, or the approval that we receive may be for a
narrower indication than we had originally sought, potentially undermining the commercial viability
of the product. Even if regulatory approvals are obtained, a marketed product is subject to
continual review, and later discovery of previously unknown problems or failure to comply with the
applicable regulatory requirements may result in restrictions on the marketing of a product or
withdrawal of the product from the market as well as possible civil or criminal sanctions. For
marketing outside the United States, we also will be subject to foreign regulatory requirements
governing human clinical trials and marketing approval for pharmaceutical products. The
requirements governing the conduct of clinical trials, product licensing, pricing, and
reimbursement vary widely from country to country.
None of our pharmaceutical products under development have been approved for marketing in the
United States or elsewhere. We may not be able to obtain regulatory approval for any such products
under development in a timely manner, if at all. Failure to obtain requisite governmental
approvals or failure to obtain approvals of the scope requested will delay or preclude us, or our
licensees or marketing partners, from marketing our products, or limit the commercial use of our
products, and thereby would have a material adverse effect on our business, financial condition,
and results of operations. See Risk Factors The results of pre-clinical trials and previous
clinical trials for our products may not be predictive of future results, and our current and
planned clinical trials may not satisfy the requirements of the FDA or other non-U.S. regulatory
authorities.
Device Development
Devices are subject to varying levels of premarket regulatory control, the most comprehensive
of which requires that a clinical evaluation be conducted before a device receives approval for
commercial distribution. The FDA classifies medical devices into one of three classes: Class I
devices are relatively simple and can be manufactured and distributed with general controls; Class
II devices are somewhat more complex and require greater scrutiny; Class III devices are new and
frequently help sustain life.
In the United States, a company generally can obtain permission to distribute a new device in
one of two ways. The first applies to any device that is substantially equivalent to a device
first marketed prior to May 1976, or to another device marketed after that date, but which was
substantially equivalent to a pre-May 1976 device. These devices are either Class I or Class II
devices. To obtain FDA permission to distribute the device, the company generally must submit a
section 510(k) submission, and receive an FDA order finding substantial equivalence to a predicate
device (pre-May 1976 or post-May 1976 device that was substantially equivalent to a pre-May 1976
device) and permitting commercial distribution of that device for its intended use. A 510(k)
submission must provide information supporting a claim of substantial equivalence to the predicate
device. If clinical data from human experience are required to support the 510(k) submission,
these data must be gathered in compliance with investigational device exemption (IDE),
regulations for investigations performed in the United States. The 510(k) process is normally used
for products of the type that the Company proposes distributing. The FDA review process for
premarket notifications submitted pursuant to section 510(k) takes, on average, about 90 days, but
it can take substantially longer if the FDA has concerns, and there is no guarantee that the FDA
will clear the device for marketing, in which case the device cannot be distributed in the United
States. There is also no guarantee that the FDA will deem the applicable device subject to the
510(k) process, as opposed to the more time-consuming, resource-intensive and problematic, pre
market approval (PMA) process described below.
The second, more comprehensive, approval process applies to a new device that is not
substantially equivalent to a pre-1976 product or that is to be used in supporting or sustaining
life or preventing impairment. These devices are normally Class III devices. For example, most
implantable devices are subject to the approval process. Two steps of FDA approval are generally
required before a company can market a product in the United States that is subject to approval, as
opposed to clearance. First, a company must comply with IDE regulations in connection with any
human clinical investigation of the device. These regulations permit a company to undertake a
clinical study of a
15
Table of Contents
non-significant risk device without formal FDA approval. Prior express FDA
approval is required if the device is a significant risk device. Second, the FDA must review the
companys PMA application, which contains, among other things, clinical information acquired under
the IDE. The FDA will approve the PMA application if it finds
there is reasonable assurance that the device is safe and effective for its intended use. The
PMA process takes substantially longer than the 510(k) process and it is conceivable that the FDA
would not agree with our assessment that a device that we propose to distribute should be a Class I
or Class II device. If that were to occur we would be required to undertake the more complex and
costly PMA process. However, for either the 510(k) or the PMA process, the FDA could require us to
run clinical trials, which would pose all of the same risks and uncertainties associated with the
clinical trials of drugs, described above.
Even when a clinical study has been approved by the FDA or deemed approved, the study is
subject to factors beyond a manufacturers control, including, but not limited to the fact that the
institutional review board at a given clinical site might not approve the study, might decline to
renew approval which is required annually, or might suspend or terminate the study before the study
has been completed. Also, the interim results of a study may not be satisfactory, leading the
sponsor to terminate or suspend the study on its own initiative or the FDA may terminate or suspend
the study. There is no assurance that a clinical study at any given site will progress as
anticipated; there may be an insufficient number of patients who qualify for the study or who agree
to participate in the study or the investigator at the site may have priorities other than the
study. Also, there can be no assurance that the clinical study will provide sufficient evidence to
assure the FDA that the product is safe and effective, a prerequisite for FDA approval of a PMA, or
substantially equivalent in terms of safety and effectiveness to a predicate device, a prerequisite
for clearance under 510(k). Even if the FDA approves or clears a device, it may limit its intended
uses in such a way that manufacturing and distributing the device may not be commercially feasible.
After clearance or approval to market is given, the FDA and foreign regulatory agencies, upon
the occurrence of certain events, are authorized under various circumstances to withdraw the
clearance or approval or require changes to a device, its manufacturing process or its labeling or
additional proof that regulatory requirements have been met.
A manufacturer of a device approved through the PMA is not permitted to make changes to the
device which affect its safety or effectiveness without first submitting a supplement application
to its PMA and obtaining FDA approval for that supplement. In some instances, the FDA may require
clinical trials to support a supplement application. A manufacturer of a device cleared through
the 510(k) process must submit another premarket notification if it intends to make a change or
modification in the device that could significantly affect the safety or effectiveness of the
device, such as a significant change or modification in design, material, chemical composition,
energy source or manufacturing process. Any change in the intended uses of a PMA device or a
510(k) device requires an approval supplement or cleared premarket notification. Exported devices
are subject to the regulatory requirements of each country to which the device is exported, as well
as certain FDA export requirements.
A company that intends to manufacture medical devices is required to register with the FDA
before it begins to manufacture the device for commercial distribution. As a result, we and any
entity that manufactures products on our behalf will be subject to periodic inspection by the FDA
for compliance with the FDAs Quality System Regulation requirements and other regulations. In the
European Community, we will be required to maintain certain International Organization for
Standardization (ISO), certifications in order to sell products and we or our manufacturers
undergo periodic inspections by notified bodies to obtain and maintain these certifications. These
regulations require us or our manufacturers to manufacture products and maintain documents in a
prescribed manner with respect to design, manufacturing, testing and control activities. Further,
we are required to comply with various FDA and other agency requirements for labeling and
promotion. The Medical Device Reporting regulations require that we provide information to the FDA
whenever there is evidence to reasonably suggest that a device may have caused or contributed to a
death or serious injury or, if a malfunction were to occur, could cause or contribute to a death or
serious injury. In addition, the FDA prohibits us from promoting a medical device for unapproved
indications.
Our instrumentation products are subject to regulation by the FDA and similar international
health authorities. We also have an obligation to adhere to the FDAs cGMP regulations.
Additionally, we are subject to periodic FDA inspections, quality control procedures, and other
detailed validation procedures. If the FDA finds deficiencies in the validation of our
manufacturing and quality control practices, they may impose restrictions on marketing specific
products until corrected.
Regulation by governmental authorities in the United States and other countries may be a
significant factor in how we develop, test, produce and market our molecular diagnostic test
products. Diagnostic tests like ours do not fall squarely within the regulatory approval process
for pharmaceutical or device products as described above, and
16
Table of Contents
the regulatory pathway is not as
clear. It is possible that diagnostic products developed by us or our collaborators will be
regulated as medical devices by the FDA and comparable agencies of other countries and require
either premarket approval (PMA) or 510(k) clearance from the FDA prior to marketing.
Nevertheless, some companies
that have successfully commercialized diagnostic tests for various conditions and disease
states have not sought clearance or approval for such tests through the traditional or PMA
processes, and have instead utilized a process involving laboratory developed tests (LDTs)
through a laboratory certified under The Clinical Laboratory Improvement Amendments of 1988
(CLIA). CLIA is a federal law that regulates clinical laboratories that perform testing on
specimens derived from humans for the purpose of providing information for diagnostic, preventative
or treatment purpose. In such instances, the CLIA lab is solely responsible for the development,
validation and commercialization of the assay. Such LDT testing is currently under the purview of
CMS and state agencies that provide oversight of the safe and effective use of LDTs. Although the
FDA has consistently claimed that it has the regulatory authority to regulate LDTs that are
validated by the developing laboratory and performed only by that laboratory, it has generally
exercised enforcement discretion in not otherwise regulating most tests developed and performed by
high complexity CLIA-certified laboratories. Recently, however, the FDA indicated that it was
reviewing the regulatory requirements that will apply to LDTs, and held a two-day public meeting on
July 19 and July 20, 2010 to obtain input from stakeholders on how it should apply its authority to
implement a reasonable, risk-based, and effective regulatory framework for LDTs. Although the FDA
did not indicate when or how those changes would be implemented, it left little doubt that the
changes are forthcoming.
Impact of Regulation
The FDA in the course of enforcing the FDCA may subject a company to various sanctions for
violating FDA regulations or provisions of the FDCA, including requiring recalls, issuing Warning
Letters, seeking to impose civil money penalties, seizing devices that the agency believes are
non-compliant, seeking to enjoin distribution of a specific type of device or other product,
seeking to revoke a clearance or approval, seeking disgorgement of profits and seeking to
criminally prosecute a company and its officers and other responsible parties.
The levels of revenues and profitability of biopharmaceutical companies may be affected by the
continuing efforts of government and third party payers to contain or reduce the costs of health
care through various means. For example, in certain foreign markets, pricing or profitability of
therapeutic and other pharmaceutical products is subject to governmental control. In the United
States, there have been, and we expect that there will continue to be, a number of federal and
state proposals to implement similar governmental control. In addition, in the United States and
elsewhere, sales of therapeutic and other pharmaceutical products are dependent in part on the
availability and adequacy of reimbursement from third party payers, such as the government or
private insurance plans. Third party payers are increasingly challenging established prices, and
new products that are more expensive than existing treatments may have difficulty finding ready
acceptance unless there is a clear therapeutic benefit. We cannot assure you that any of our
products will be considered cost effective, or that reimbursement will be available or sufficient
to allow us to sell them competitively and profitably.
Our instrumentation products are subject to regulation by the FDA and similar international
health authorities. We also have an obligation to adhere to the FDAs cGMP regulations.
Additionally, we are subject to periodic FDA inspections, quality control procedures, and other
detailed validation procedures. If the FDA finds deficiencies in the validation of our
manufacturing and quality control practices, they may impose restrictions on marketing specific
products until corrected.
Anti-Kickback Laws
We are also subject to various federal, state, and international laws pertaining to health
care fraud and abuse, including anti-kickback laws and false claims laws. Anti-kickback laws
make it illegal to solicit, offer, receive or pay any remuneration in exchange for, or to induce,
the referral of business, including the purchase or prescription of a particular drug or the use of
a service or device. Federal and state false claims laws prohibit anyone from knowingly and
willingly presenting, or causing to be presented for payment to third-party payors (including
Medicare and Medicaid), claims for reimbursed drugs or services that are false or fraudulent,
claims for items or services not provided as claimed, or claims for medically unnecessary items or
services. If the government were to allege against or convict us of violating these laws, there
could be a material adverse effect on us, including our stock price. Even an unsuccessful
challenge could cause adverse publicity and be costly to respond to, which could have a materially
adverse effect on our business, results of operations and financial condition. We will consult
counsel concerning the potential application of these and other laws to our business and our sales,
marketing and other activities and will make good faith efforts to comply with them. However,
given their broad reach and the
17
Table of Contents
increasing attention given by law enforcement authorities, we
cannot assure you that some of our activities will not be challenged or deemed to violate some of
these laws.
Foreign Corrupt Practices Act
We are also subject to the U.S. Foreign Corrupt Practices Act (FCPA), which prohibits
corporations and individuals from paying, offering to pay, or authorizing the payment of anything
of value to any foreign government official, government staff member, political party, or political
candidate in an attempt to obtain or retain business or to otherwise influence a person working in
an official capacity. The FCPA also requires public companies to make and keep books and records
that accurately and fairly reflect their transactions and to devise and maintain an adequate system
of internal accounting controls. Our international activities create the risk of unauthorized
payments or offers of payments by our employees, consultants, sales agents or distributors, even
though they may not always be subject to our control. We discourage these practices by our
employees and agents. However, our existing safeguards and any future improvements may prove to be
less than effective, and our employees, consultants, sales agents or distributors may engage in
conduct for which we might be held responsible. Any failure by us to adopt appropriate compliance
procedures and ensure that our employees and agents comply with the FCPA and applicable laws and
regulations in foreign jurisdictions could result in substantial penalties or restrictions on our
ability to conduct business in certain foreign jurisdictions.
MANUFACTURING AND QUALITY
Other than our facility in Guadalajara, Mexico, we currently have no pharmaceutical
manufacturing facilities. We have entered into agreements with various third parties for the
formulation and manufacture of our pharmaceutical clinical supplies. These suppliers and their
manufacturing facilities must comply with FDA regulations, current good laboratory practices
(cGLPs) and current good manufacturing practices (cGMPs). We plan to outsource the
manufacturing and formulation of our clinical supplies.
We have an instrumentation manufacturing facility in Hialeah, Florida, which predominantly
performs high level assembly for our instrumentation products. Certain of our products components
and optical subsystems are produced by sub-contracted vendors that specialize in optical device
manufacturing.
We are committed to providing high quality products to our customers, and we plan to meet this
commitment by working diligently to continue implementing updated and improved quality systems and
concepts throughout our organization.
SALES & MARKETING
We currently do not have pharmaceutical or diagnostics sales or marketing personnel in the
United States and have limited personnel in Chile and Mexico. In order to commercialize any
pharmaceutical or diagnostic products that are approved for commercial sale, we must either build a
sales and marketing infrastructure or collaborate with third parties with sales and marketing
experience.
Our instrumentation division has offices in the United States and the United Kingdom and a
distributor network that currently covers more than 50 countries. Our strategy is to increase
sales of existing products through expansion of our sales channel in the United States and to
provide additional marketing resources to our international distributor network.
SERVICE & SUPPORT
We currently offer service and telephone support for all of our marketed instrumentation
products. Warranties are given on all products against defects of labor and material. Extended
Service Contracts are available for purchase. Product repairs are performed onsite at our Hialeah
facility.
EMPLOYEES
As of December 31, 2010, we had 220 full-time employees worldwide. None of our employees are
represented by a collective bargaining agreement.
18
Table of Contents
Code of Ethics
We have adopted a Code of Business Conduct and Ethics. We require all employees, including
our principal executive officer and principal accounting officer and other senior officers and our
employee directors, to read and to adhere to the Code of Business Conduct and Ethics in discharging
their work-related responsibilities. Employees are required to report any conduct that they
believe in good faith to be an actual or apparent violation of the Code of Business Conduct and
Ethics. The Code of Business Conduct and Ethics is available on our website at
http://www.OPKO.com.
Available Information
We make available free of charge on or through our web site, at www.opko.com, our Annual Reports
on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments
to those reports as soon as reasonably practicable after such material is electronically filed
with the SEC. Additionally, the public may read and copy any materials we file with the SEC
at the SECs Public Reference Room at 100 F Street, NE, Room 1580, Washington, D.C., 20549.
Information regarding operation of the Public Reference Room is available by calling the SEC
at 1-800-SEC-0330. Information that we file with the SEC is also available at the SECs Web
site at www.sec.gov.
19
Table of Contents
ITEM 1A. RISK FACTORS.
You should carefully consider the risks described below, as well as other information
contained in this report, including the consolidated financial statements and the notes thereto and
Managements Discussion and Analysis of Financial Condition and Results of Operations. The
occurrence of any of the events discussed below could significantly and adversely affect our
business, prospects, results of operations, financial condition, and cash flows.
RISKS RELATED TO OUR BUSINESS
We have a history of operating losses and we do not expect to become profitable in the near future.
We are a healthcare company with a limited operating history. We are not profitable and have
incurred losses since our inception. We do not anticipate that we will generate revenue from the
sale of proprietary pharmaceutical products or our molecular diagnostic products for some time and
we have generated limited revenue from our pharmaceutical operations in Chile and Mexico and from
our instrumentation business. We have not yet submitted any pharmaceutical products or molecular
diagnostic products for marketing approval or clearance by regulatory authorities and we do not
currently have rights to any pharmaceutical product candidates that have been approved for
marketing, other than those products sold by our Chilean and Mexican subsidiaries. We continue to
incur research and development and general and administrative expenses related to our operations
and, to date, we have devoted most of our financial resources to research and development,
including our pre-clinical development activities and clinical trials. We expect to continue to
incur losses from our operations for the foreseeable future, and we expect these losses to increase
as we continue our research activities and conduct development of, and seek regulatory approvals
and clearances for, our product candidates, and prepare for and begin to commercialize any approved
or cleared products. If our product candidates fail in clinical trials or do not gain regulatory
approval or clearance, or if our product candidates do not achieve market acceptance, we may never
become profitable. In addition, if we are required by the U.S. Food and Drug Administration
(FDA), to perform studies in addition to those we currently anticipate, our expenses will
increase beyond expectations and the timing of any potential product approval may be delayed. Even
if we achieve profitability in the future, we may not be able to sustain profitability in
subsequent periods.
Our technologies are in an early stage of development and are unproven.
The effectiveness of our technologies is not well-known in, or accepted generally by, the
clinical medical community. There can be no assurance that we will be able to successfully employ
our technologies as therapeutic, diagnostic, or preventative solutions for any disease or
condition. Our failure to establish the efficacy or safety of our technologies would have a
material adverse effect on our business.
In addition, we have a limited operating history. Our operations to date have been primarily
limited to organizing and staffing our company, developing our technology, and undertaking
pre-clinical studies and clinical trials of our product candidates. We have not yet obtained
regulatory approvals for any of our pharmaceutical product or molecular diagnostic candidates.
Consequently, any predictions you make about our future success or viability may not be as accurate
as they could be if we had a longer operating history.
Our product research and development activities may not result in commercially viable products.
Most of our product candidates, including our molecular diagnostic products and vaccine
technologies, are in the early stages of development and are prone to the risks of failure inherent
in drug, diagnostic, and medical device product development. These risks further include the
possibility that such products would:
| | be found to be ineffective, unreliable, or otherwise inadequate or otherwise fail to receive regulatory approval; | ||
| | be difficult or impossible to manufacture on a commercial scale; | ||
| | be uneconomical to market or otherwise not be effectively marketed; | ||
| | fail to be successfully commercialized if adequate reimbursement from government health administration authorities, private health insurers, and other organizations for the costs of these products is unavailable; |
20
Table of Contents
| | be impossible to commercialize because they infringe on the proprietary rights of others or compete with products marketed by others that are superior; or | ||
| | fail to be commercialized prior to the successful marketing of similar products by competitors. |
The results of pre-clinical trials and previous clinical trials for our products may not be
predictive of future results, and our current and planned clinical trials may not satisfy the
requirements of the FDA or other non-U.S. regulatory authorities.
Positive results from pre-clinical studies and early clinical trial experience should not be
relied upon as evidence that later-stage or large-scale clinical trials will succeed. We may be
required to demonstrate with substantial evidence through well-controlled clinical trials that our
product candidates either (i) are safe and effective for use in a diverse population of their
intended uses or (ii) with respect to Class I or Class II devices only, are substantially
equivalent in terms of safety and effectiveness to devices that are already marketed under section
510(k) of the Food, Drug and Cosmetic Act. Success in early clinical trials does not mean that
future clinical trials will be successful because product candidates in later-stage clinical trials
may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and other
non-U.S. regulatory authorities despite having progressed through initial clinical trials.
Further, our drug candidates may not be approved or cleared even if they achieve their primary
endpoints in Phase III clinical trials or registration trials. In addition our device candidates,
as well as our molecular diagnostic candidates, may not be approved or cleared, as the case may be,
even though clinical or other data are, in our view, adequate to support a device or diagnostic
test approval or clearance. The FDA or other non-U.S. regulatory authorities may disagree with our
trial design and our interpretation of data from pre-clinical studies and clinical trials. In
addition, any of these regulatory authorities may change requirements for the approval or clearance
of a product candidate even after reviewing and providing comment on a protocol for a pivotal
clinical trial that has the potential to result in FDA and other non-U.S. regulatory authorities
approval. Any of these regulatory authorities may also approve or clear a product candidate for
fewer or more limited indications or uses than we request or may grant approval or clearance
contingent on the performance of costly post-marketing clinical trials. The FDA or other non-U.S.
regulatory authorities may not approve the labeling claims necessary or desirable for the
successful commercialization of our product candidates.
The results of our clinical trials may show that our product candidates may cause undesirable
side effects, which could interrupt, delay or halt clinical trials, resulting in the denial of
regulatory approval by the FDA and other non-U.S. regulatory authorities.
In light of widely publicized events concerning the safety risk of certain drug products,
regulatory authorities, members of Congress, the Government Accounting Office, medical
professionals, and the general public have raised concerns about potential drug safety issues.
These events have resulted in the withdrawal of drug products, revisions to drug labeling that
further limit use of the drug products, and establishment of risk management programs that may, for
instance, restrict distribution of drug products. The increased attention to drug safety issues
may result in a more cautious approach by the FDA to clinical trials. Data from clinical trials
may receive greater scrutiny with respect to safety, which may make the FDA or other regulatory
authorities more likely to terminate clinical trials before completion, or require longer or
additional clinical trials that may result in substantial additional expense and a delay or failure
in obtaining approval or approval for a more limited indication than originally sought.
We will require substantial additional funding, which may not be available to us on acceptable
terms, or at all.
We are advancing and intend to continue to advance multiple product candidates through
clinical and pre-clinical development. On March 14, 2011, we issued 27,000,000 shares of our
common stock in an underwritten public offering at a price of $3.75 per share. The net proceeds
received in the offering were approximately $96.4 million. We believe we have sufficient cash and
cash equivalents on hand or available to us through lines of credit to meet our anticipated cash
requirements for operations and debt service beyond the next 12 months. We have based this
estimate on assumptions that may prove to be wrong or subject to change, and we may be required to
use our available capital resources sooner than we currently expect or curtail aspects of our
operations in order to preserve our capital. Because of the numerous risks and uncertainties
associated with the development and commercialization of our product candidates, we are unable to
estimate the amounts of increased capital outlays and operating expenditures associated with our
current and anticipated clinical trials. Our future capital requirements will depend on a number
of factors, including the continued progress of our research and development of product candidates,
the timing and outcome of clinical trials and regulatory approvals, the costs involved in
preparing, filing,
21
Table of Contents
prosecuting, maintaining, defending, and enforcing patent claims and other intellectual
property rights, the status of competitive products, the availability of financing, and our success
in developing markets for our product candidates.
Until we can generate a sufficient amount of product revenue to finance our cash requirements,
which we may never do, we expect to finance future cash needs primarily through public or private
equity offerings, debt financings, or strategic collaborations. Our ability to obtain additional
capital may depend on prevailing economic conditions and financial, business and other factors
beyond our control. Disruptions in the United States and global financial markets may adversely
impact the availability and cost of credit, as well as our ability to raise money in the capital
markets. Economic conditions have been, and continue to be, volatile. Continued instability in
these market conditions may limit our ability to replace, in a timely manner, maturing liabilities
and access the capital necessary to fund and grow our business. There can be no assurance that
additional capital will be available to us on acceptable terms, or at all, which could adversely
impact our business, results of operations, liquidity, capital resources and financial condition.
If we are not able to secure additional funding when needed, we may have to delay, reduce the scope
of, or eliminate one or more of our clinical trials or research and development programs. To the
extent that we raise additional funds by issuing equity securities, our stockholders may experience
additional significant dilution, and debt financing, if available, may involve restrictive
covenants. To the extent that we raise additional funds through collaboration and licensing
arrangements, it may be necessary to relinquish some rights to our technologies or our product
candidates or grant licenses on terms that may not be favorable to us. We may seek to access the
public or private capital markets whenever conditions are favorable, even if we do not have an
immediate need for additional capital at that time.
Our business is substantially dependant on our ability to develop, launch and generate revenue from
our molecular diagnostic program.
Our business is substantially dependant on our ability to develop and launch simple diagnostic
tests based on our molecular diagnostics platform for Alzheimers disease, cancers and other
conditions for which we are developing tests. We are committing significant research and
development resources to the development of such diagnostic tests, and there is no guarantee that
we will be able to successfully launch these or other diagnostic tests on anticipated timelines or
at all. We have limited experience in developing, manufacturing, selling, marketing or
distributing tests based on the molecular diagnostic platform. If we are not able to successfully
develop, market or sell diagnostic tests we develop for any reason, including the failure to obtain
any required regulatory approvals, we will not generate any revenue from the sale of such tests.
Even if we are able to develop effective diagnostic tests for sale in the marketplace, a number of
factors could impact our ability to sell such tests or generate any significant revenue from the
sale of such tests, including without limitation:
| | our ability to establish and maintain adequate infrastructure to support the commercial launch and sale of our diagnostic tests ourselves or through a CLIA certified laboratory, including establishing adequate laboratory space, information technology infrastructure, sample collection and tracking systems, electronic ordering and reporting systems and other infrastructure and hiring adequate laboratory and other personnel; | ||
| | the success of the validation studies for our diagnostic tests under development and our ability to publish study results in peer-reviewed journals; | ||
| | the availability of alternative and competing tests or products and technological innovations or other advances in medicine that cause our technologies to be less competitive; | ||
| | the accuracy rates of such tests, including rates of false-negatives and/or false-positives; | ||
| | concerns regarding the safety or effectiveness or clinical utility of our diagnostic tests; | ||
| | changes in the regulatory environment affecting health care and health care providers, including changes in laws regulating laboratory testing and/or device manufacturers; | ||
| | the extent and success of our sales and marketing efforts and ability to drive adoption of our diagnostic tests; | ||
| | coverage and reimbursement levels by government payors and private insurers; |
22
Table of Contents
| | pricing pressures and changes in third-party payor reimbursement policies; and | ||
| | intellectual property rights held by others or others infringing our intellectual property rights. |
If our competitors develop and market products that are more effective, safer or less expensive
than our future product candidates, our commercial opportunities will be negatively impacted.
The pharmaceutical, molecular diagnostic, and instrumentation industries are highly
competitive and require an ongoing, extensive search for technological innovation. Numerous
companies, including major pharmaceutical companies, specialty pharmaceutical companies and
specialized biotechnology companies, are engaged in the development, manufacture and marketing of
pharmaceutical products competitive with those that we intend to commercialize ourselves and
through our partners, including without limitation, Merck, Genentech, Allergan, Alcon Laboratories,
Novartis, Alnylam, Regeneron, and QLT. Competitors to our molecular diagnostics business are many
and include major diagnostic companies, reference laboratories, molecular diagnostic firms,
universities and research institutions. Most of these companies have substantially greater
financial and other resources, larger research and development staffs and more extensive marketing
and manufacturing organizations than ours. Large pharmaceutical companies, in particular, have
extensive experience in clinical testing and in obtaining regulatory approvals or clearances for
drugs or medical devices. These companies also have significantly greater research and marketing
capabilities than we do. Compared to us, many of our potential competitors have substantially
greater capital resources, development resources, including personnel and technology, clinical
trial experience, regulatory experience, expertise in prosecution of intellectual property rights,
manufacturing and distribution experience, and sales and marketing experience.
We believe that our ability to successfully compete will depend on, among other things:
| | the results of our clinical trials; | ||
| | our ability to recruit and enroll patients for our clinical trials; | ||
| | the efficacy, safety and reliability of our product candidates; | ||
| | the speed at which we develop our product candidates; | ||
| | our ability to design and successfully execute appropriate clinical trials; | ||
| | the timing and scope of regulatory approvals or clearances; | ||
| | our ability to commercialize and market any of our product candidates that may receive regulatory approval or clearance; | ||
| | appropriate coverage and adequate levels of reimbursement under private and governmental health insurance plans, including Medicare; | ||
| | our ability to protect intellectual property rights related to our products; | ||
| | our ability to have our partners manufacture and sell commercial quantities of any approved products to the market; and | ||
| | acceptance of future product candidates by physicians and other health care providers. |
If our competitors market products that are more effective, safer, easier to use or less
expensive than our future product candidates, if any, or that reach the market sooner than our
future product candidates, if any, we may not achieve commercial success. In addition, the
biopharmaceutical, molecular diagnostic, and medical device industries are characterized by rapid
technological change. Because our research approach integrates many technologies, it may be
difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at
the forefront of technological change, we may be unable to compete effectively. Technological
advances or products developed by our competitors may render our technologies or product candidates
obsolete or less competitive.
23
Table of Contents
Our product development activities could be delayed or stopped.
We do not know whether our current or planned pre-clinical and clinical studies will be
completed on schedule, or at all. Furthermore, we cannot guarantee that our planned pre-clinical
and clinical studies will begin on time or at all. The commencement of our planned clinical trials
could be substantially delayed or prevented by several factors, including:
| | a limited number of, and competition for, suitable patients with the particular types of disease required for enrollment in our clinical trials or that otherwise meet the protocols inclusion criteria and do not meet any of the exclusion criteria; | ||
| | a limited number of, and competition for, suitable sites to conduct our clinical trials; | ||
| | delay or failure to obtain FDA or other non-U.S. regulatory authorities approval or agreement to commence a clinical trial; | ||
| | delay or failure to obtain sufficient supplies of the product candidate for our clinical trials; | ||
| | requirements to provide the drugs or medical devices required in our clinical trial protocols or clinical trials at no cost or cost, which may require significant expenditures that we are unable or unwilling to make; | ||
| | delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or investigators; and | ||
| | delay or failure to obtain institutional review board (IRB) approval to conduct or renew a clinical trial at a prospective site. |
The completion of our clinical trials could also be substantially delayed or prevented by
several factors, including:
| | slower than expected rates of patient recruitment and enrollment; | ||
| | failure of patients to complete the clinical trial; | ||
| | unforeseen safety issues; | ||
| | lack of efficacy evidenced during clinical trials; | ||
| | termination of our clinical trials by one or more clinical trial sites; | ||
| | inability or unwillingness of patients or medical investigators to follow our clinical trial protocols; and | ||
| | inability to monitor patients adequately during or after treatment. |
Our clinical trials may be suspended or terminated at any time by the FDA, other regulatory
authorities, the IRB for any given site, or us. Additionally, changes in regulatory requirements
and guidance may occur and we may need to amend clinical trial protocols to reflect these changes
with appropriate regulatory authorities. Amendments may require us to resubmit our clinical trial
protocols to IRBs for re-examination, which may impact the costs, timing, or successful completion
of a clinical trial. Any failure or significant delay in commencing or completing clinical trials
for our product candidates could materially harm our results of operations and financial condition,
as well as the commercial prospects for our product candidates.
The regulatory approval process is expensive, time consuming and uncertain and may prevent us or
our collaboration partners from obtaining approvals for the commercialization of some or all of our
product candidates.
The research, testing, manufacturing, labeling, approval, selling, marketing, and distribution
of drug products or medical devices are subject to extensive regulation by the FDA and other
non-U.S. regulatory authorities, which regulations differ from country to country. In general, we
are not permitted to market our product candidates in the United States until we receive approval
of a new drug application (NDA), a clearance letter under the premarket
24
Table of Contents
notification process, or 510(k) process, or an approval of a pre-market approval (PMA) from
the FDA. We have not submitted a NDA or PMA application or premarket notification, nor have we
received marketing approval or clearance for any of our proprietary pharmaceutical or diagnostic
product candidates. Obtaining approval of a NDA or PMA can be a lengthy, expensive, and uncertain
process. With respect to medical devices, while the FDA reviews and clears a premarket
notification in as little as three months, there is no guarantee that our products will qualify for
this more expeditious regulatory process, which is reserved for Class I and II devices, nor is
there any assurance that even if a device is reviewed under the 510(k) process that the FDA will
review it expeditiously or determine that the device is substantially equivalent to a lawfully
marketed non-PMA device. If the FDA fails to make this finding, then we cannot market the device.
In lieu of acting on a premarket notification, the FDA may seek additional information or
additional data which would further delay our ability to market the product. Furthermore, we are
not permitted to make changes to a device approved through the PMA or 510(k) which affects the
safety or efficacy of the device without first submitting a supplement application to the PMA and
obtaining FDA approval or cleared premarket notification for that supplement. In some cases, the
FDA may require clinical trials to support a supplement application. In addition, failure to
comply with FDA, non-U.S. regulatory authorities, or other applicable United States and non-U.S.
regulatory requirements may, either before or after product approval or clearance, if any, subject
our company to administrative or judicially imposed sanctions, including, but not limited to the
following:
| | restrictions on the products, manufacturers, or manufacturing process; | ||
| | adverse inspectional observations (Form 483), warning letters, or non-warning letters incorporating inspectional observations; | ||
| | civil and criminal penalties; | ||
| | injunctions; | ||
| | suspension or withdrawal of regulatory approvals or clearances; | ||
| | product seizures, detentions, or import bans; | ||
| | voluntary or mandatory product recalls and publicity requirements; | ||
| | total or partial suspension of production; | ||
| | imposition of restrictions on operations, including costly new manufacturing requirements; and | ||
| | refusal to approve or clear pending NDAs or supplements to approved NDAs, applications or pre-market notifications. |
Regulatory approval of an NDA or NDA supplement, PMA, PMA supplement or clearance pursuant to
a pre-market notification is not guaranteed, and the approval or clearance process, as the case may
be, is expensive and may, especially in the case of an NDA or PMA application, take several years.
The FDA also has substantial discretion in the drug and medical device approval and clearance
process. Despite the time and expense exerted, failure can occur at any stage, and we could
encounter problems that cause us to abandon clinical trials or to repeat or perform additional
pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials
that will be required for FDA approval or clearance varies depending on the drug or medical device
candidate, the disease or condition that the drug or medical device candidate is designed to
address, and the regulations applicable to any particular drug or medical device candidate. The
FDA can delay, limit or deny approval or clearance of a drug or medical device candidate for many
reasons, including:
| | a drug candidate may not be deemed safe or effective; | ||
| | a medical device candidate may not be deemed to be substantially equivalent to a lawfully marketed non-PMA device, in the case of a premarket notification; | ||
| | the FDA may not find the data from pre-clinical studies and clinical trials sufficient; | ||
| | the FDA may not approve our or our third-party manufacturers processes or facilities; or | ||
| | the FDA may change its approval or clearance policies or adopt new regulations. |
25
Table of Contents
Regulation by governmental authorities in the United States and other countries may be a
significant factor in how we develop, test, produce and market our molecular diagnostic test
products. Diagnostic tests like ours do not fall squarely within the regulatory approval process
for pharmaceutical or device products as described above, and the regulatory pathway is not as
clear. It is possible that diagnostic products developed by us or our collaborators will be
regulated as medical devices by the FDA and comparable agencies of other countries and require
either PMA or 510(k) clearance from the FDA prior to marketing. Some companies that have
successfully commercialized diagnostic tests for various conditions and disease states have not
sought clearance or approval for such tests through the traditional 510(k) or PMA processes, and
have instead utilized a process involving laboratory developed tests (LDTs) through a laboratory
certified under The Clinical Laboratory Improvement Amendments of 1988 (CLIA). CLIA is a federal
law that regulates clinical laboratories that perform testing on specimens derived from humans for
the purpose of providing information for diagnostic, preventative or treatment purpose. In such
instances, the CLIA lab is solely responsible for the development, validation and commercialization
of the assay. Such LDT testing is currently under the purview of CMS and state agencies that
provide oversight of the safe and effective use of LDTs. Although the FDA has consistently claimed
that is has the regulatory authority to regulate LDTs that are validated by the developing
laboratory and performed only by that laboratory, it has generally exercised enforcement discretion
in not otherwise regulating most tests developed and performed by high complexity CLIA-certified
laboratories. Recently, however, the FDA indicated that it was reviewing the regulatory
requirements that will apply to LDTs, and held a two-day public meeting on July 19 and July 20,
2010 to obtain input from stakeholders on how it should apply its authority to implement a
reasonable, risk-based, and effective regulatory framework for LDTs. Although the FDA did not
indicate when or how those changes would be implemented, it left little doubt that the changes are
forthcoming.
Our product candidates may have undesirable side effects and cause our approved drugs to be taken
off the market.
If a product candidate receives marketing approval and we or others later identify undesirable
side effects caused by such products:
| | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication, or field alerts to physicians and pharmacies; | ||
| | regulatory authorities may withdraw their approval of the product and require us to take our approved drug off the market; | ||
| | we may be required to change the way the product is administered, conduct additional clinical trials, or change the labeling of the product; | ||
| | we may have limitations on how we promote our drugs; | ||
| | sales of products may decrease significantly; | ||
| | we may be subject to litigation or product liability claims; and | ||
| | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the
affected product or could substantially increase our commercialization costs and expenses, which in
turn could delay or prevent us from generating significant revenues from its sale.
Our inability to address quality control issues in a timely manner could delay the production and
sale of our instrumentation products.
We are committed to providing high quality products to our customers, and we plan to meet this
commitment by working diligently to continue implementing updated and improved quality systems and
concepts throughout our organization. We cannot assure you that we will not have quality control
issues in the future, which may result in warning letters and citations from the FDA. If we
receive any warning letters from the FDA in the future, there can be no assurances regarding the
length of time or cost it will take us to resolve such quality issues to our satisfaction and to
the satisfaction of the FDA. If our remedial actions are not satisfactory to the FDA, we may have
to devote
26
Table of Contents
additional financial and human resources to our efforts, and the FDA may take further
regulatory actions against us including, but not limited to, assessing civil monetary penalties or
imposing a consent decree on us, which could result in further regulatory constraints, including
the governance of our quality system by a third party. Our inability to resolve these issues or
the taking of further regulatory action by the FDA may weaken our competitive position and have a
material adverse effect on our business, results of operations and financial condition.
We manufacture products in Mexico through our Mexican subsidiary. Any quality control issues
at our Mexican facility may weaken our competitive position and have a material adverse effect on
our business results of operations and financial condition.
We may not meet regulatory quality standards applicable to our manufacturing and quality processes,
which could have an adverse effect on our business, results of operations and financial condition.
As a medical device manufacturer, we are required to register with the FDA and are subject to
periodic inspection by the FDA for compliance with its Quality System Regulation (QSR)
requirements, which require manufacturers of medical devices to adhere to certain regulations,
including testing, quality control and documentation procedures. Compliance with applicable
regulatory requirements is subject to continual review and is monitored rigorously through periodic
inspections by the FDA. In addition, most international jurisdictions have adopted regulatory
approval and periodic renewal requirements for medical devices, and we must comply with these
requirements in order to market our products in these jurisdictions. In the European Community, we
are required to maintain certain ISO certifications in order to sell our products and must undergo
periodic inspections by notified bodies to obtain and maintain these certifications. Further, some
emerging markets rely on the FDAs Certificate for Foreign Government (CFG) in lieu of their own
regulatory approval requirements. Our, or our manufacturers failure to meet QSR ISO, or any other
regulatory requirements or industry standards could delay production of our products and lead to
fines, difficulties in obtaining regulatory clearances, recalls or other consequences, which could,
in turn, have a material adverse effect on our business, results of operations, and our financial
condition.
Even if we obtain marketing approvals or clearances for our product candidates, the terms of
approvals and ongoing regulation of our products may limit how we manufacture and market our
product candidates, which could materially impair our ability to generate anticipated revenues.
Once regulatory approval has been granted to market a product, the approved or cleared product
and its manufacturer are subject to continual review. Any approved or cleared product may only be
promoted for its indicated uses. In addition, if the FDA or other non-U.S. regulatory authorities
approve any of our product candidates for marketing, the labeling, packaging, adverse event
reporting, storage, advertising, and promotion for the product will be subject to extensive
regulatory requirements. We and the manufacturers of our products are also required to comply with
current Good Manufacturing Practices (cGMP) regulations or the FDAs QSR regulations, which
include requirements relating to quality control and quality assurance as well as the corresponding
maintenance of records and documentation. Moreover, device manufacturers are required to report
adverse events by filing Medical Device Reports with the FDA, which reports are publicly available.
Further, regulatory agencies must approve manufacturing facilities before they can be used to
manufacture our products, and these facilities are subject to ongoing regulatory inspection. If we
fail to comply with the regulatory requirements of the FDA and other non-U.S. regulatory
authorities, or if previously unknown problems with our products, manufacturers, or manufacturing
processes are discovered, we could be subject to administrative or judicially imposed sanctions.
Furthermore, any limitation on indicated uses for a product candidate or our ability to manufacture
and promote a product candidate could significantly and adversely affect our business, results of
operations, and financial condition.
In addition, the FDA and other non-U.S. regulatory authorities may change their policies and
additional regulations may be enacted that could prevent or delay marketing approval or clearance
of our product candidates. We cannot predict the likelihood, nature or extent of government
regulation that may arise from future legislation or administrative action, either in the United
States or abroad. If we are not able to maintain regulatory compliance, we would likely not be
permitted to market our future product candidates and we may not achieve or sustain profitability,
which would materially impair our ability to generate anticipated revenues.
27
Table of Contents
Even if we receive regulatory approval or clearance to market our product candidates, the market
may not be receptive to our products.
Even if our product candidates obtain marketing approval or clearance, our products may not
gain market acceptance among physicians, patients, health care payors and/or the medical community.
We believe that the degree of market acceptance will depend on a number of factors, including:
| | timing of market introduction of competitive products; | ||
| | safety and efficacy of our product compared to other products; | ||
| | prevalence and severity of any side effects; | ||
| | potential advantages or disadvantages over alternative treatments; | ||
| | strength of marketing and distribution support; | ||
| | price of our products, both in absolute terms and relative to alternative treatments; | ||
| | availability of coverage and reimbursement from government and other third-party payors; | ||
| | potential product liability claims; | ||
| | limitations or warnings contained in a products regulatory authority-approved labeling; and | ||
| | changes in the standard of care for the targeted indications for any of our product candidates, which could reduce the marketing impact of any claims that we could make following applicable regulatory authority approval. |
In addition, our efforts to educate the medical community and health care payors on the
benefits of our product candidates may require significant resources and may never be successful.
If our products do not gain market acceptance, it would have a material adverse effect on our
business, results of operations, and financial condition.
If our future product candidates are not covered and eligible for reimbursement from government and
third party payors, we may not be able to generate significant revenue or achieve or sustain
profitability.
The coverage and reimbursement status of newly approved or cleared drugs, diagnostic tests or
medical devices is uncertain, and failure of our pharmaceutical and diagnostic products and
procedures using our medical devices to be adequately covered by insurance and eligible for
adequate reimbursement could limit our ability to market any future product candidates we may
develop and decrease our ability to generate revenue from any of our existing and future product
candidates that may be approved or cleared.
There is significant uncertainty related to the third-party coverage and reimbursement of
newly approved or cleared drugs, diagnostic products, or medical devices. Many medical devices are
not directly covered by insurance; instead, the procedure using the device is subject to a coverage
determination by the insurer. The commercial success of our existing and future product candidates
in both domestic and international markets will depend in part on the availability of coverage and
adequate reimbursement from third-party payors, including government payors, such as the Medicare
and Medicaid programs, managed care organizations, and other third-party payors. The government
and other third-party payors are increasingly attempting to contain health care costs by limiting
both insurance coverage and the level of reimbursement for new drugs, diagnostic tests, or devices
and, as a result, they may not cover or provide adequate payment for our product candidates. These
payors may conclude that our product candidates are less safe, less effective, or less
cost-effective than existing or later-introduced products. These payors may also conclude that the
overall cost of the procedure using one of our devices exceeds the overall cost of the competing
procedure using another type of device, and third-party payors may not approve our product
candidates for insurance coverage and adequate reimbursement. The failure to obtain coverage and
adequate or any reimbursement for our product candidates, or health care cost containment
initiatives that limit or restrict reimbursement for our product candidates, may reduce any future
product revenue. Even though a drug (not administered by a physician) may be approved by the FDA,
this does not mean that a Prescription Drug Plan (PDP), a private insurer operating under
Medicare part D, will list that drug on its formulary or will set a reimbursement level. PDPs are
not required to make every FDA-approved drug available on their formularies. If
28
Table of Contents
our drug products are not listed on sufficient number of PDP formularies or if the PDPs
levels of reimbursement are inadequate, the Companys business, results of operations, and
financial condition could be materially adversely affected.
If we fail to attract and retain key management and scientific personnel, we may be unable to
successfully develop or commercialize our product candidates.
We will need to expand and effectively manage our managerial, operational, financial,
development, and other resources in order to successfully pursue our research, development, and
commercialization efforts for our product candidates. Our success depends on our continued ability
to attract, retain, and motivate highly qualified management and pre-clinical and clinical
personnel. The loss of the services or support of any of our senior management, particularly Dr.
Phillip Frost, our Chairman of the Board and Chief Executive Officer, could delay or prevent the
development and commercialization of our product candidates. We do not maintain key man
insurance policies on the lives of any of our employees. We will need to hire additional personnel
as we continue to expand our research and development activities and build a sales and marketing
function.
We have scientific and clinical advisors who assist us in formulating our research,
development, and clinical strategies. These advisors are not our employees and may have
commitments to, or consulting or advisory contracts with, other entities that may limit their
availability to us. In addition, these advisors may have arrangements with other companies to
assist those companies in developing products or technologies that may compete with ours.
We may not be able to attract or retain qualified management and scientific personnel in the
future due to the intense competition for qualified personnel among biotechnology, pharmaceutical,
medical device, and other similar businesses. If we are unable to attract and retain the necessary
personnel to accomplish our business objectives, we may experience constraints that will impede
significantly the achievement of our research and development objectives, our ability to raise
additional capital and our ability to implement our business strategy, which will adversely affect
our business, results of operations and financial condition. In particular, if we lose any members
of our senior management team, we may not be able to find suitable replacements in a timely fashion
or at all and our business may be harmed as a result.
As we evolve from a company primarily involved in development to a company also involved in
commercialization, we may encounter difficulties in managing our growth and expanding our
operations successfully.
As we advance our product candidates through clinical trials, research, and development we
will need to expand our development, regulatory, manufacturing, marketing, and sales capabilities
or contracts with third parties to provide these capabilities for us. As our operations expand, we
expect that we will need to manage additional relationships with such third parties, as well as
additional collaborators and suppliers. Maintaining these relationships and managing our future
growth will impose significant added responsibilities on members of our management. We must be
able to: manage our development efforts effectively; manage our clinical trials effectively; hire,
train and integrate additional management, development, administrative and sales and marketing
personnel; improve our managerial, development, operational and finance systems; implement and
manage an effective marketing strategy; and expand our facilities, all of which may impose a strain
on our administrative and operational infrastructure.
Furthermore, we may acquire additional businesses, products or product candidates that
complement or augment our existing business. Integrating any newly acquired business or product
could be expensive and time-consuming. We may not be able to integrate any acquired business or
product successfully or operate any acquired business profitably. Our future financial performance
will depend, in part, on our ability to manage any future growth effectively and our ability to
integrate any acquired businesses. We may not be able to accomplish these tasks, and our failure
to accomplish any of them could prevent us from successfully growing our company, which would have
a material adverse effect on our business, results of operations and financial condition.
If we fail to acquire and develop other products or product candidates at all or on commercially
reasonable terms, we may be unable to diversify or grow our business.
We intend to continue to rely on acquisitions and in-licensing as the source of our products
and product candidates for development and commercialization. The success of this strategy depends
upon our ability to identify, select, and acquire pharmaceutical and diagnostic products, drug
delivery technologies, and medical device product candidates. Proposing, negotiating, and
implementing an economically viable product acquisition or license
29
Table of Contents
is a lengthy and complex process. We compete for partnering arrangements and license
agreements with pharmaceutical, biotechnology and medical device companies, and academic research
institutions. Our competitors may have stronger relationships with third parties with whom we are
interested in collaborating and/or may have more established histories of developing and
commercializing products. Most of our competitors also have substantially greater financial and
other resources than us. As a result, our competitors may have a competitive advantage in entering
into partnering arrangements with such third parties, as such partnering arrangements are often
decided in an auction process in which the highest bidder wins. In addition, even if we find
promising product candidates, and generate interest in a partnering or strategic arrangement to
acquire such product candidates, we may not be able to acquire rights to additional product
candidates or approved products on terms that we find acceptable, or at all.
We expect that any product candidate to which we acquire rights will require additional
development efforts prior to commercial sale, including extensive clinical testing and approval or
clearance by the FDA and other non-U.S. regulatory authorities. All product candidates are subject
to the risks of failure inherent in pharmaceutical, diagnostic test or medical device product
development, including the possibility that the product candidate will not be shown to be
sufficiently safe and effective for approval by regulatory authorities. Even if the product
candidates are approved or cleared for marketing, we cannot be sure that they would be capable of
economically feasible production or commercial success. If we fail to acquire or develop other
product candidates that are capable of economically feasible production and commercial success, our
business, results of operations and financial condition and cash flows may be materially adversely
affected.
We have no experience or capability manufacturing large clinical-scale or commercial-scale products
and have no pharmaceutical manufacturing facility other than our facility in Guadalajara, Mexico;
we therefore rely on third parties to manufacture and supply our pharmaceutical product candidates.
If our manufacturing partners are unable to produce our products in the amounts that we
require, we may not be able to establish a contract and obtain a sufficient alternative supply from
another supplier on a timely basis and in the quantities we require. We expect to continue to
depend on third-party contract manufacturers for the foreseeable future.
Our product candidates require precise, high quality manufacturing. Any of our contract
manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and other
non-U.S. regulatory authorities to ensure strict compliance with QSR regulations for devices or
cGMPs for drugs, and other applicable government regulations and corresponding standards relating
to matters such as testing, quality control, and documentation procedures. If our contract
manufacturers fail to achieve and maintain high manufacturing standards in compliance with QSR or
cGMPs, we may experience manufacturing errors resulting in patient injury or death, product recalls
or withdrawals, delays or interruptions of production or failures in product testing or delivery,
delay or prevention of filing or approval of marketing applications for our products, cost
overruns, or other problems that could seriously harm our business.
Any performance failure on the part of our contract manufacturers could delay clinical
development or regulatory approval or clearance of our product candidates or commercialization of
our future product candidates, depriving us of potential product revenue and resulting in
additional losses. In addition, our dependence on a third party for manufacturing may adversely
affect our future profit margins. Our ability to replace an existing manufacturer may be difficult
because the number of potential manufacturers is limited and the FDA must approve any replacement
manufacturer before it can begin manufacturing our product candidates. Such approval would result
in additional non-clinical testing and compliance inspections. It may be difficult or impossible
for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or
at all.
We currently have limited marketing staff and no pharmaceutical or diagnostic sales or distribution
capabilities in the United States. If we are unable to develop our sales, marketing and
distribution capability on our own or through collaborations with marketing partners, we will not
be successful in commercializing our pharmaceutical or diagnostic product candidates in the United
States.
We currently have no pharmaceutical or diagnostic test marketing, sales or distribution
capabilities other than through our Mexican and Chilean subsidiaries for sales in those countries.
If our pharmaceutical product candidates are approved, we intend to establish our sales and
marketing organization with technical expertise and supporting distribution capabilities to
commercialize our product candidates, which will be expensive and time-consuming. Any failure or
delay in the development of any of our internal sales, marketing, and distribution capabilities
would adversely impact the commercialization of our products. With respect to our existing and
future pharmaceutical
30
Table of Contents
product candidates, we may choose to collaborate with third parties that have direct sales
forces and established distribution systems, either to augment our own sales force and distribution
systems or in lieu of our own sales force and distribution systems. To the extent that we enter
into co-promotion or other licensing arrangements, our product revenue is likely to be lower than
if we directly marketed or sold our products. In addition, any revenue we receive will depend in
whole or in part upon the efforts of such third parties, which may not be successful and are
generally not within our control. If we are unable to enter into such arrangements on acceptable
terms or at all, we may not be able to successfully commercialize our existing and future product
candidates. If we are not successful in commercializing our existing and future product
candidates, either on our own or through collaborations with one or more third parties, our future
product revenue will suffer and we may incur significant additional losses.
Independent clinical investigators and contract research organizations that we engage to conduct
our clinical trials may not be diligent, careful or timely.
We depend on independent clinical investigators to conduct our clinical trials. Contract
research organizations may also assist us in the collection and analysis of data. These
investigators and contract research organizations will not be our employees, and we will not be
able to control, other than by contract, the amount of resources, including time, that they devote
to products that we develop. If independent investigators fail to devote sufficient resources to
the development of product candidates or clinical trials, or if their performance is substandard,
it will delay the marketing approval or clearance and commercialization of any products that we
develop. Further, the FDA requires that we comply with standards, commonly referred to as good
clinical practice, for conducting, recording and reporting clinical trials to assure that data and
reported results are credible and accurate and that the rights, integrity, and confidentiality of
trial subjects are protected. If our independent clinical investigators and contract research
organizations fail to comply with good clinical practice, the results of our clinical trials could
be called into question and the clinical development of our product candidates could be delayed.
Failure of clinical investigators or contract research organizations to meet their obligations to
us or comply with federal regulations and good clinical practice procedures could adversely affect
the clinical development of our product candidates and harm our business, results of operations,
and financial condition.
The success of our business may be dependent on the actions of our collaborative partners.
We expect to enter into collaborative arrangements with established multi-national
pharmaceutical and medical device companies, which will finance or otherwise assist in the
development, manufacture and marketing of products incorporating our technology. We anticipate
deriving some revenues from research and development fees, license fees, milestone payments, and
royalties from collaborative partners. Our prospects, therefore, may depend to some extent upon
our ability to attract and retain collaborative partners and to develop technologies and products
that meet the requirements of prospective collaborative partners. In addition, our collaborative
partners may have the right to abandon research projects, guide strategy regarding prosecution of
relevant patent applications and terminate applicable agreements, including funding obligations,
prior to or upon the expiration of the agreed-upon research terms. There can be no assurance that
we will be successful in establishing collaborative arrangements on acceptable terms or at all,
that collaborative partners will not terminate funding before completion of projects, that our
collaborative arrangements will result in successful product commercialization, or that we will
derive any revenues from such arrangements. To the extent that we are unable to develop and
maintain collaborative arrangements, we would need substantial additional capital to undertake
research, development, and commercialization activities on our own.
Our license agreement with TESARO, Inc. is important to our business. If TESARO does not
successfully develop and commercialize rolapitant, our business could be adversely affected.
In December 2010, we exclusively out-licensed the development, manufacture and
commercialization of rolapitant to TESARO, Inc., an oncology-focused biopharmaceutical company
founded by executives with a demonstrated track record in launching successful products for the
CINV market. TESARO is initially pursuing development and commercialization of rolapitant for
CINV. Under the terms of the license, we are eligible to receive payments of up to $121.0 million,
including an up-front payment of $6.0 million we received in December 2010, and additional payments
based upon net sales and achievement of specified regulatory and commercialization milestones. In
addition, TESARO will pay us double digit tiered royalties on sales of licensed product. Further,
we will share with TESARO future profits from the commercialization of licensed products in Japan,
and we will have an option to market the products in Latin America. If TESARO fails to
successfully develop and commercialize rolapitant, we may not receive any milestone or royalty
payments under the license agreement, which could have a material adverse impact on our financial
condition.
31
Table of Contents
If we are unable to obtain and enforce patent protection for our products, our business could be
materially harmed.
Our success depends, in part, on our ability to protect proprietary methods and technologies
that we develop or license under the patent and other intellectual property laws of the United
States and other countries, so that we can prevent others from unlawfully using our inventions and
proprietary information. However, we may not hold proprietary rights to some patents required for
us to commercialize our product candidates. Because certain U.S. patent applications are
confidential until patents issue, such as applications filed prior to November 29, 2000, or
applications filed after such date for which nonpublication has been requested, third parties may
have filed patent applications for technology covered by our pending patent applications without
our being aware of those applications, and our patent applications may not have priority over those
applications. For this and other reasons, we or our third-party collaborators may be unable to
secure desired patent rights, thereby losing desired exclusivity. If licenses are not available to
us on acceptable terms, we may not be able to market the affected products or conduct the desired
activities, unless we challenge the validity, enforceability, or infringement of the third-party
patent or otherwise circumvent the third-party patent.
Our strategy depends on our ability to rapidly identify and seek patent protection for our
discoveries. In addition, we will rely on third-party collaborators to file patent applications
relating to proprietary technology that we develop jointly during certain collaborations. The
process of obtaining patent protection is expensive and time-consuming. If our present or future
collaborators fail to file and prosecute all necessary and desirable patent applications at a
reasonable cost and in a timely manner, our business will be adversely affected. Despite our
efforts and the efforts of our collaborators to protect our proprietary rights, unauthorized
parties may be able to obtain and use information that we regard as proprietary.
The issuance of a patent does not guarantee that it is valid or enforceable. Any patents we
have obtained, or obtain in the future, may be challenged, invalidated, unenforceable, or
circumvented. Moreover, the U.S. Patent and Trademark Office (USPTO) may commence interference
proceedings involving our patents or patent applications. In addition, court decisions may
introduce uncertainty in the enforceability or scope of patents owned by biotechnology,
pharmaceutical, and medical device companies. Any challenge to, finding of unenforceability or
invalidation or circumvention of, our patents or patent applications would be costly, would require
significant time and attention of our management, and could have a material adverse effect on our
business, results of operations and financial condition.
Our pending patent applications may not result in issued patents. The patent position of
pharmaceutical, biotechnology, and medical device companies, including ours, is generally uncertain
and involves complex legal and factual considerations. The standards that the U.S. Patent and
Trademark Office and its foreign counterparts use to grant patents are not always applied
predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the
subject matter and scope of claims granted or allowable in pharmaceutical, biotechnology, or
medical device patents. Accordingly, we do not know the degree of future protection for our
proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to
others. The legal systems of certain countries do not favor the aggressive enforcement of patents,
and the laws of foreign countries may not protect our rights to the same extent as the laws of the
United States. Therefore, the enforceability or scope of our owned or licensed patents in the
United States or in foreign countries cannot be predicted with certainty, and, as a result, any
patents that we own or license may not provide sufficient protection against competitors. We may
not be able to obtain or maintain patent protection for our pending patent applications, those we
may file in the future, or those we may license from third parties, including the University of
Pennsylvania, the University of Texas Southwestern Medical Center and Academia Sinica.
While we believe that our patent rights are enforceable, we cannot assure you that any patents
that have issued, that may issue, or that may be licensed to us will be enforceable or valid, or
will not expire prior to the commercialization of our product candidates, thus allowing others to
more effectively compete with us. Therefore, any patents that we own or license may not adequately
protect our product candidates or our future products, which could have a material adverse effect
on our business, results of operations, and financial condition.
32
Table of Contents
We do not have an exclusive arrangement in place with The Scripps Research Institute or Dr. Tom
Kodadek with respect to technology or intellectual property that may be material to our business.
If any such technology or intellectual property is developed by The Scripps Research Institute or
its employees, including Dr. Kodadek, and we are unable to license such technology or intellectual
property, or such technology or intellectual property is licensed to or acquired by other parties,
our business and competitive position may be materially harmed.
Our success depends, in part, on our ability to develop and protect proprietary methods,
products and technologies. Dr. Tom Kodadek, who currently serves as our Director of Chemistry &
Molecular Biology is a staff member and employee of The Scripps Research Institute (TSRI), a
private, non-profit research organization. Dr. Kodadek, as our consultant, supervises our research
and development efforts with respect to our molecular diagnostics program, and the creation of
intellectual property that is important to our business. We have entered into consulting
arrangements with TSRI and Dr. Kodadek, with respect to Dr. Kodadeks services to us. We have the
right to intellectual property resulting from Dr. Kodadeks services to us under these
arrangements. However, we do not have an exclusive arrangement with Dr. Kodadek or TSRI, and Dr.
Kodadek also provides services to TSRI and other third parties and may provide services to other
third parties in the future. We do not have any rights to any technology or intellectual property
that may be developed by TSRI and its employees, including Dr. Kodadek, outside of these
arrangements. If TSRI or its employees, including Dr. Kodadek, develops technology or intellectual
property that is material to our business and we are unable to license such technology or
intellectual property on favorable terms, if at all, or such technology or intellectual property is
licensed to or acquired by other parties, our business and competitive position may be harmed.
If we are unable to protect the confidentiality of our proprietary information and know-how, the
value of our technology and products could be adversely affected.
In addition to patent protection, we also rely on other proprietary rights, including
protection of trade secrets, know-how, and confidential and proprietary information. To maintain
the confidentiality of trade secrets and proprietary information, we will seek to enter into
confidentiality agreements with our employees, consultants, and collaborators upon the commencement
of their relationships with us. These agreements generally require that all confidential
information developed by the individual or made known to the individual by us during the course of
the individuals relationship with us be kept confidential and not disclosed to third parties. Our
agreements with employees also generally provide that any inventions conceived by the individual in
the course of rendering services to us shall be our exclusive property.
However, we may not obtain these agreements in all circumstances, and individuals with whom we
have these agreements may not comply with their terms. In the event of unauthorized use or
disclosure of our trade secrets or proprietary information, these agreements, even if obtained, may
not provide meaningful protection, particularly for our trade secrets or other confidential
information. To the extent that our employees, consultants, or contractors use technology or
know-how owned by third parties in their work for us, disputes may arise between us and those third
parties as to the rights in related inventions.
Adequate remedies may not exist in the event of unauthorized use or disclosure of our
confidential information. The disclosure of our trade secrets would impair our competitive
position and may materially harm our business, financial condition, and results of operations.
We will rely heavily on licenses from third parties. Failure to comply with the provisions of
these licenses could result in the loss of our rights under the license agreements.
Many of the patents and patent applications in our patent portfolio are not owned by us, but
are licensed from third parties. For example, we rely on technology licensed from the University
of Pennsylvania, UT Southwestern, and Academia Sinica, among others. Such license agreements give
us rights for the commercial exploitation of the patents resulting from the respective patent
applications, subject to certain provisions of the license agreements. Failure to comply with
these provisions could result in the loss of our rights under these license agreements. Our
inability to rely on these patents and patent applications, which are the basis of our technology,
would have a material adverse effect on our business, results of operations and financial
condition.
33
Table of Contents
We license patent rights to certain of our technology from third-party owners. If such owners do
not properly maintain or enforce the patents underlying such licenses, our competitive position and
business prospects will be harmed.
We have obtained licenses from, among others, the University of Pennsylvania, UT Southwestern,
and Academia Sinica, that are necessary or useful for our business. In addition, we intend to
enter into additional licenses of third-party intellectual property in the future. Although our
goal is to obtain exclusivity in our licensing transactions, we cannot guarantee that no third
parties will step forward and assert inventorship or ownership in our in-licensed patents. In some
cases, we may rely on the assurances of our licensors that all ownership rights have been secured
and that all necessary agreements are intact or forthcoming.
Our success will depend in part on our ability or the ability of our licensors to obtain,
maintain, and enforce patent protection for our licensed intellectual property and, in particular,
those patents to which we have secured exclusive rights in our field. We or our licensors may not
successfully prosecute the patent applications which are licensed to us. Even if patents issue in
respect of these patent applications, we or our licensors may fail to maintain these patents, may
determine not to pursue litigation against other companies that are infringing these patents, or
may pursue such litigation less aggressively than we would. Without protection for the
intellectual property we have licensed, other companies might be able to offer substantially
identical products for sale, which could adversely affect our competitive business position and
harm our business, results of operations and financial condition.
Some jurisdictions may require us, or those from whom we license patents, to grant licenses to
third parties. Such compulsory licenses could be extended to include some of our product
candidates, which may limit our potential revenue opportunities.
Many countries, including certain countries in Europe, have compulsory licensing laws under
which a patent owner may be compelled to grant licenses to third parties. In addition, most
countries limit the enforceability of patents against government agencies or government
contractors. In these countries, the patent owner may be limited to monetary relief from an
infringement and may be unable to enjoin infringement, which could materially diminish the value of
the patent. If we or those from who we license patents are required to issue compulsory licenses,
it could materially adversely affect our business, results of operation and financial condition.
Our commercial success depends significantly on our ability to operate without infringing the
patents and other proprietary rights of third parties.
Other entities may have or obtain patents or proprietary rights that could limit our ability
to develop, manufacture, use, sell, offer for sale or import products, or impair our competitive
position. In addition, other entities may have or obtain patents or proprietary rights that cover
our current research and preclinical studies. While there are statutory exemptions to patent
infringement for those who are using third party patented technology in the process of pursuing FDA
regulatory approval, the U.S. case law pertaining to such exemptions changes over time. Lawsuits
involving such exemptions are very fact intensive and it is currently unclear under U.S. case law
whether preclinical studies would always qualify for such an exemption, and whether such exemptions
would apply to research tools. To the extent that our current research and preclinical studies may
be covered by the patent rights of others, the risk of suit may continue after such patents expire
because the statute of limitations for patent infringement runs for six years. To the extent that
a third party develops and patents technology that covers our products, we may be required to
obtain licenses to that technology, which licenses may not be available or may not be available on
commercially reasonable terms, if at all. If licenses are not available to us on acceptable terms,
we will not be able to market the affected products or conduct the desired activities, unless we
challenge the validity, enforceability or infringement of the third-party patent, or circumvent the
third-party patent, which would be costly and would require significant time and attention of our
management. Third parties may have or obtain by license or assignment valid and enforceable
patents or proprietary rights that could block us from developing products using our technology.
Our failure to obtain a license to any technology that we require may materially harm our business,
financial condition, and results of operations.
If we become involved in patent litigation or other proceedings related to a determination of
rights, we could incur substantial costs and expenses, substantial liability for damages or be
required to stop our product development and commercialization efforts.
Third parties may sue us for infringing their patent rights. Likewise, we may need to resort
to litigation to enforce a patent issued or licensed to us or to determine the scope and validity
of proprietary rights of others. In
34
Table of Contents
addition, a third-party may claim that we have improperly obtained or used its confidential or
proprietary information. Furthermore, in connection with our third-party license agreements, we
generally have agreed to indemnify the licensor for costs incurred in connection with litigation
relating to intellectual property rights. The cost to us of any litigation or other proceeding
relating to intellectual property rights, even if resolved in our favor, could be substantial, and
the litigation would divert our managements efforts. Some of our competitors may be able to
sustain the costs of complex patent litigation more effectively than we can because they have
substantially greater resources. Uncertainties resulting from the initiation and continuation of
any litigation could limit our ability to continue our operations. Our involvement in patent
litigation and other proceedings could have a material adverse effect on our business, results of
operations, and financial condition.
If any parties successfully claim that our creation or use of proprietary technologies
infringes upon their intellectual property rights, we might be forced to pay damages, potentially
including treble damages, if we are found to have willfully infringed on such parties patent
rights. In addition to any damages we might have to pay, a court could require us to stop the
infringing activity or obtain a license. Any license required under any patent may not be made
available on commercially acceptable terms, if at all. In addition, such licenses are likely to be
non-exclusive and, therefore, our competitors may have access to the same technology licensed to
us. If we fail to obtain a required license and are unable to design around a patent, we may be
unable to effectively market some of our technology and products, which could limit our ability to
generate revenues or achieve profitability and possibly prevent us from generating revenue
sufficient to sustain our operations.
Adverse results in material litigation matters or governmental inquiries could have a material
adverse effect upon our business and financial condition.
We may from time to time become subject in the ordinary course of business to material legal
action related to, among other things, intellectual property disputes, professional liability,
contractual and employee-related matters, as well as inquiries from governmental agencies and
Medicare or Medicaid carriers requesting comment and information on allegations of billing
irregularities and other matters that are brought to their attention through billing audits, third
parties or other sources. The health care industry is subject to substantial federal and state
government regulation and audit. Legal actions could result in substantial monetary damages as
well as damage to the Companys reputation with customers, which could have a material adverse
effect upon our results of operations and financial position.
Medicare legislation and future legislative or regulatory reform of the health care system may
affect our ability to sell our products profitably.
In the United States, there have been a number of legislative and regulatory initiatives, at
both the federal and state government levels, to change the healthcare system in ways that, if
approved, could affect our ability to sell our products profitably. While many of the proposed
policy changes require congressional approval to implement, we cannot assure you that reimbursement
payments under governmental and private third party payer programs will remain at levels comparable
to present levels or will be sufficient to cover the costs allocable to patients eligible for
reimbursement under these programs. Any changes that lower reimbursement rates under Medicare,
Medicaid or private pay programs could negatively affect our business.
In addition, there are efforts underway to attempt the passage of significant healthcare
reform legislation. Any such health care reform may have an adverse effect on our business through
decreasing funds available to our customers and to us. Limitations or restrictions on Medicare and
Medicaid payments to our customers could adversely impact the liquidity of our customers, resulting
in their inability to pay us, or to timely pay us, for our products and services. This inability
could have a material adverse effect on our financial position, results of operations and
liquidity.
We are unable to predict what additional legislation or regulation, if any, relating to the
health care industry or third-party coverage and reimbursement may be enacted in the future or what
effect such legislation or regulation would have on our business.
35
Table of Contents
RISKS RELATED TO INTERNATIONAL OPERATIONS
Failure to obtain regulatory approval outside the United States will prevent us from marketing our
product candidates abroad.
We intend to market certain of our existing and future product candidates in non-U.S. markets.
In order to market our existing and future product candidates in the European Union and many other
non-U.S. jurisdictions, we must obtain separate regulatory approvals. We have had limited
interactions with non-U.S. regulatory authorities, the approval procedures vary among countries and
can involve additional testing, and the time required to obtain approval may differ from that
required to obtain FDA approval or clearance. Approval or clearance by the FDA does not ensure
approval by regulatory authorities in other countries, and approval by one or more non-U.S.
regulatory authority does not ensure approval by other regulatory authorities in other countries or
by the FDA. The non-U.S. regulatory approval process may include all of the risks associated with
obtaining FDA approval or clearance. We may not obtain non-U.S. regulatory approvals on a timely
basis, if at all. We may not be able to file for non-U.S. regulatory approvals and may not receive
necessary approvals to commercialize our existing and future product candidates in any market,
which would have a material adverse effect on our business, results of operations and financial
condition.
Non-U.S. governments often impose strict price controls, which may adversely affect our future
profitability.
We intend to seek approval to market certain of our existing and future product candidates in
both the United States and in non-U.S. jurisdictions. If we obtain approval in one or more
non-U.S. jurisdictions, we will be subject to rules and regulations in those jurisdictions relating
to our product. In some countries, particularly countries of the European Union, each of which has
developed its own rules and regulations, pricing is subject to governmental control. In these
countries, pricing negotiations with governmental authorities can take considerable time after the
receipt of marketing approval for a drug or medical device candidate. To obtain reimbursement or
pricing approval in some countries, we may be required to conduct a clinical trial that compares
the cost-effectiveness of our existing and future product candidates to other available products.
If reimbursement of our future product candidates is unavailable or limited in scope or amount, or
if pricing is set at unsatisfactory levels, we may be unable to generate revenues and achieve or
sustain profitability, which would have a material adverse effect on our business, results of
operations and financial condition.
We may be exposed to liabilities under the Foreign Corrupt Practices Act, and any determination
that we violated the Foreign Corrupt Practices Act could have a material adverse effect on our
business.
We are subject to the Foreign Corrupt Practice Act (FCPA) and other laws that prohibit U.S.
companies or their agents and employees from providing anything of value to a foreign official or
political party for the purposes of influencing any act or decision of these individuals in their
official capacity to help obtain or retain business, direct business to any person or corporate
entity or obtain any unfair advantage. We have operations and agreements with third parties and we
generate sales internationally. Our international activities create the risk of unauthorized and
illegal payments or offers of payments by our employees, consultants, sales agents or distributors,
even though they may not always be subject to our control. We discourage these practices by our
employees and agents. However, our existing safeguards and any future improvements may prove to be
less than effective, and our employees, consultants, sales agents or distributors may engage in
conduct for which we might be held responsible. Any failure by us to adopt appropriate compliance
procedures and ensure that our employees and agents comply with the FCPA and applicable laws and
regulations in foreign jurisdictions could result in substantial penalties or restrictions on our
ability to conduct business in certain foreign jurisdictions.
Violations of the FCPA may result in severe criminal or civil sanctions, and we may be subject
to other liabilities, which could negatively affect our business, operating results and financial
condition. In addition, the U.S. government may seek to hold our Company liable for successor
liability FCPA violations committed by companies in which we invest or that we acquire.
We are subject to risks associated with doing business globally.
Our operations, both within and outside the United States, are subject to risks inherent in
conducting business globally and under the laws, regulations and customs of various jurisdictions
and geographies. These risks include fluctuations in currency exchange rates, changes in exchange
controls, loss of business in government tenders that are held annually in many cases,
nationalization, increasingly complex labor environments, expropriation and other governmental
actions, changes in taxation, including legislative changes in U.S. and international taxation of
income
36
Table of Contents
earned outside of the United States, importation limitations, export control restrictions,
violations of U.S. or local laws, including the U.S. Foreign Corrupt Practices Act, dependence on a
few government entities as customers, pricing restrictions, economic destabilization, political and
economic instability, disruption or destruction in a significant geographic region due to the
location of manufacturing facilities, distribution facilities or customers regardless of cause,
including war, terrorism, riot, civil insurrection or social unrest, or natural or man-made
disasters, including famine, flood, fire, earthquake, storm or disease. Failure to comply with the
laws and regulations that affect our global operations, could have an adverse effect on our
business, financial condition or results of operations.
RISKS RELATED TO ACQUISITIONS
Acquisitions, investments and strategic alliances that we have made or may make in the future
may use significant resources, result in disruptions to our business or distractions of our
management, may not proceed as planned, and could expose us to unforeseen liabilities. We intend
to continue to expand our business through the acquisition of, investments in and strategic
alliances with companies, technologies, products, and services. Acquisitions, investments and
strategic alliances involve a number of special problems and risks, including, but not limited to:
| | difficulty integrating acquired technologies, products, services, operations, and personnel with the existing businesses; | ||
| | diversion of managements attention in connection with both negotiating the acquisitions and integrating the businesses; | ||
| | strain on managerial and operational resources as management tries to oversee larger operations; | ||
| | difficulty implementing and maintaining effective internal control over financial reporting at businesses that we acquire, particularly if they are not located near our existing operations; | ||
| | exposure to unforeseen liabilities of acquired companies; | ||
| | potential costly and time-consuming litigation, including stockholder lawsuits; | ||
| | potential issuance of securities to equity holders of the company being acquired with rights that are superior to the rights of holders of our common stock, or which may have a dilutive effect on our stockholders; | ||
| | the need to incur additional debt or use cash; and | ||
| | the requirement to record potentially significant additional future operating costs for the amortization of intangible assets. |
As a result of these or other problems and risks, businesses we acquire may not produce the
revenues, earnings, or business synergies that we anticipated, and acquired products, services, or
technologies might not perform as we expected. As a result, we may incur higher costs and realize
lower revenues than we had anticipated. We may not be able to successfully address these problems
and we cannot assure you that the acquisitions will be successfully identified and completed or
that, if acquisitions are completed, the acquired businesses, products, services, or technologies
will generate sufficient revenue to offset the associated costs or other negative effects on our
business.
Any of these risks can be greater if an acquisition is large relative to our size. Failure to
manage effectively our growth through acquisitions could adversely affect our growth prospects,
business, results of operations, financial condition and cash flows.
Funding may not be available for us to continue to make acquisitions, investments and strategic
alliances in order to grow our business.
We have made and anticipate that we may continue to make acquisitions, investments and
strategic alliances with complementary businesses, technologies, products and services to expand
our business. Our growth plans rely, in part, on the successful completion of future acquisitions.
At any particular time, we may need to raise substantial additional capital or to issue additional
equity to finance such acquisitions, investments, and strategic alliances.
37
Table of Contents
There is no assurance that we will be able to secure additional funding on acceptable terms,
or at all, or obtain the stockholder approvals necessary to issue additional equity to finance such
acquisitions, investments, and strategic alliances. If we are unsuccessful in obtaining the
financing, our business would be adversely impacted.
RISKS RELATED TO OWNERSHIP OF OUR COMMON STOCK
The market price of our common stock may fluctuate significantly.
The market price of our common stock may fluctuate significantly in response to numerous
factors, some of which are beyond our control, such as:
| | the announcement of new products or product enhancements by us or our competitors; | ||
| | results of our clinical trials and other development efforts; | ||
| | developments concerning intellectual property rights and regulatory approvals; | ||
| | variations in our and our competitors results of operations; | ||
| | changes in earnings estimates or recommendations by securities analysts, if our common stock is covered by analysts; | ||
| | developments in the biotechnology, pharmaceutical, and medical device industry; | ||
| | the results of product liability or intellectual property lawsuits; | ||
| | future issuances of common stock or other securities, including debt; | ||
| | sales of stock by our officers, directors or affiliates; | ||
| | the addition or departure of key personnel; | ||
| | announcements by us or our competitors of acquisitions, investments, or strategic alliances; and | ||
| | general market conditions and other factors, including factors unrelated to our operating performance. |
Further, the stock market in general, and the market for biotechnology, pharmaceutical,
diagnostic, and medical device companies in particular, has recently experienced extreme price and
volume fluctuations. Continued market fluctuations could result in extreme volatility in the price
of our common stock, which could cause a decline in the value of our common stock.
Trading of our common stock is limited and restrictions imposed by securities regulation and
certain lockup agreements may further reduce our trading, making it difficult for our stockholders
to sell shares.
Our common stock began trading on the American Stock Exchange, now known as the NYSE Amex, in
June 2007. To date, the liquidity of our common stock is limited, not only in terms of the number
of shares that can be bought and sold at a given price, but also through delays in the timing of
transactions and changes in security analyst and media coverage, if at all.
A substantial percentage of the outstanding shares of our common stock (including outstanding
shares of our preferred stock on an as converted basis) are restricted securities and/or are
subject to lockup agreements which limit sales for a period of time. These factors may result in
lower prices for our common stock than might otherwise be obtained and could also result in a
larger spread between the bid and ask prices for our common stock. In addition, without a large
float, our common stock is less liquid than the stock of companies with broader public ownership
and, as a result, the trading prices of our common stock may be more volatile. In the absence of
an active public trading market, an investor may be unable to liquidate his investment in our
common stock. Further, the limited liquidity could be an indication that the trading price is not
reflective of the actual fair market value of our common stock. Trading of a relatively small
volume of our common stock may have a greater impact on the trading price of our stock than would
be the case if our public float were larger.
38
Table of Contents
Future sales of our common stock could reduce our stock price.
Some or all of the restricted shares of our common stock issued to former stockholders of
Froptix and Acuity in connection with the acquisition or held by other of our stockholders may be
offered from time to time in the open market pursuant to an effective registration statement, or
beginning April 2, 2008, pursuant to Rule 144. In addition, as described herein, a substantial
number of our shares of common stock were subject to lockup agreements which expired on March 27,
2009. We have also issued or agreed to issue a substantial number of securities in private
placement transactions with two year lockup restrictions expiring in each of December 2009, August
2010, and February 2011. In connection with our Series D Preferred Stock offering, shares were
issued with a three year lockup restriction that expires in September 2012. Sales of a substantial
number of shares of our common stock in the public market pursuant to Rule 144 or after the lockup
agreements lapse, or the perception that such sales could occur, could adversely affect the price
of our common stock.
Directors, executive officers, principal stockholders and affiliated entities own a majority of our
capital stock, and they may make decisions that you do not consider to be in the best interests of
our stockholders.
As of March 8, 2010, our directors, executive officers, principal stockholders, and affiliated
entities beneficially owned, in the aggregate a majority of our outstanding voting securities.
Frost Gamma Investments Trust (Gamma Trust), of which Phillip Frost, M.D., the Companys Chairman
and CEO, is the sole trustee, is deemed to beneficially own in the aggregate approximately 49% of
the Companys common stock as of March 8, 2010. As a result, Dr. Frost acting alone or with other
members of management, would have the ability to control the election of our Board of Directors,
the adoption or amendment of provisions in the Companys Certificate of Incorporation, the approval
of mergers and other significant corporate transactions, and the outcome of issues requiring
approval by our stockholders. This concentration of ownership may also have the effect of delaying
or preventing a change in control of our company that may be favored by other stockholders. This
could prevent transactions in which stockholders might otherwise recover a premium for their shares
over current market prices.
Failure to maintain effective internal controls in accordance with Section 404 of the
Sarbanes-Oxley Act could have a material adverse effect on our business and operating results. In
addition, current and potential stockholders could lose confidence in our financial reporting,
which could have a material adverse effect on the price of our common stock.
Section 404 of the Sarbanes-Oxley Act of 2002 requires annual management assessments of the
effectiveness of our internal control over financial reporting and a report by our independent
registered public accounting firm on the effectiveness of internal control over financial reporting
as of December 31, 2010. We are required to report, among other things, control deficiencies that
constitute material weaknesses or changes in internal control that, or that are reasonably likely
to, materially affect internal control over financial reporting. A material weakness is a
significant deficiency or combination of significant deficiencies that results in more than a
remote likelihood that a material misstatement of the annual or interim financial statements will
not be prevented or detected. In connection with our November 2010 restatement of our previously
issued consolidated financial statements as of and for the three and nine months ended September
30, 2009, and as of and for the year ended December 31, 2009, we determined that a deficiency in
controls relating to the accounting for a beneficial conversion feature on, and the classification
of, convertible preferred stock existed as of the previous assessment date and further concluded
that such a deficiency represented a material weakness as of December 31, 2009. As a result, we
concluded that our internal control over financial reporting was not effective as of December 31,
2009. Effective internal controls are necessary for us to provide reliable financial reports and
effectively prevent fraud. If we cannot provide reliable financial reports or prevent fraud, our
results of operation could be harmed. Our failure to maintain the effective internal control over
financial reporting could cause the cost related to remediation to increase and could cause our
stock price to decline. In addition, we may not be able to accurately report our financial
results, may be subject to regulatory sanction, and investors may lose confidence in our financial
statements. We can provide no assurance that we will at all times in the future be able to report
that our internal control is effective.
Compliance with changing regulations concerning corporate governance and public disclosure may
result in additional expenses.
There have been changing laws, regulations, and standards relating to corporate governance and
public disclosure, including the Sarbanes-Oxley Act of 2002, new regulations promulgated by the
Securities and Exchange Commission and rules promulgated by the NYSE Amex, the other national
securities exchanges and the NASDAQ. These new or changed laws, regulations, and standards are
subject to varying interpretations in many cases due to their lack of specificity, and, as a
result, their application in practice may evolve over time as new guidance is provided by
regulatory and governing bodies, which could result in continuing uncertainty regarding compliance
39
Table of Contents
matters and higher costs necessitated by ongoing revisions to disclosure and governance
practices. As a result, our efforts to comply with evolving laws, regulations, and standards are
likely to continue to result in increased general and administrative expenses and a diversion of
management time and attention from revenue-generating activities to compliance activities. Our
board members, Chief Executive Officer, Chief Financial Officer, and Principal Accounting Officer
could face an increased risk of personal liability in connection with the performance of their
duties. As a result, we may have difficulty attracting and retaining qualified board members and
executive officers, which could harm our business. If our efforts to comply with new or changed
laws, regulations, and standards differ from the activities intended by regulatory or governing
bodies, we could be subject to liability under applicable laws or our reputation may be harmed,
which could materially adversely affect our business, results of operations and financial
condition.
The conversion of shares of our preferred stock or exercise of warrants we have issued may result
in dilution to the holders of our common stock and cause the price of our common stock to decline.
As of December 31, 2010, we had 897,438 outstanding shares of Series A Preferred Stock and
1,209,677 outstanding shares of Series D Preferred Stock, which were convertible as of such date
into 897,438 and 12,096,770 shares of our common stock, respectively. In addition, as of December
31, 2010, we had outstanding warrants to purchase 29,194,162 shares of our common stock. The
conversion of outstanding shares of our Series A Preferred Stock and Series D Preferred Stock and
the exercise of warrants may result in substantial dilution to our existing stockholders and could
have a material adverse effect on our stock price. The possibility of the issuance of shares of
our common stock upon the conversion of our preferred stock or the exercise of warrants could cause
our stock price to decline as well. In addition, our preferred stockholders have dividend priority
and liquidation preferences over shares of our common stock. Thus, the rights of the holders of
common stock are and will be subject to, and may be adversely affected by, the rights of the
holders of our preferred stock. As of December 31, 2010, our Series A Preferred Stock and Series D
Preferred Stock had liquidation preferences of $2.5 million and $33.0 million, respectively.
ITEM 1B. UNRESOLVED STAFF COMMENTS.
None.
ITEM 2. PROPERTIES.
Our principal corporate office is located at 4400 Biscayne Blvd, Miami, Florida. We lease
this space from Frost Real Estate Holdings, LLC, an entity which is controlled by Dr. Phillip
Frost, our Chairman of the Board and Chief Executive Officer. Pursuant to the lease agreement with
Frost Real Estate Holdings, we lease approximately 8,300 square feet, which encompasses space for
our corporate offices, administrative services, project management and pharmacology. The lease is
for a five-year term and currently requires annual rent of approximately $0.3 million which amount
increases by approximately 4.5% per year.
We lease approximately 10,000 square feet of space in Hialeah, Florida from an entity
controlled by Dr. Phillip Frost, our Chairman and Chief Executive Officer, and Dr. Jane Hsiao, our
Vice Chairman and Chief Technical Officer, to house manufacturing and service operations for our
ophthalmic instrumentation business. We also lease facilities at Scripps Research Institute
Jupiter, which is where our molecular diagnostics research and development is based. We maintain a
research and development branch office in the United Kingdom at the University of Kent. OPKO
Chile, our Chilean subsidiary, leases office space in Santiago, Chile, and through our Mexican
subsidiaries, we own a manufacturing facility, laboratory and office space consisting of
approximately 38,000 square feet.
ITEM 3. LEGAL PROCEEDINGS.
None.
ITEM 4. (REMOVED AND RESERVED).
40
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANTS COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Our common stock is traded publicly on the NYSE Amex (formerly the American Stock Exchange)
under the symbol OPK. The following table sets forth, for the periods indicated, the high and
low sales prices per share of our common stock during each of the quarters set forth below as
reported on the NYSE Amex:
| High | Low | |||||||
2010 |
||||||||
First Quarter |
$ | 2.07 | $ | 1.63 | ||||
Second Quarter |
2.55 | 1.87 | ||||||
Third Quarter |
2.60 | 2.07 | ||||||
Fourth Quarter |
3.88 | 2.23 | ||||||
2009 |
||||||||
First Quarter |
$ | 1.70 | $ | 0.60 | ||||
Second Quarter |
1.87 | 0.98 | ||||||
Third Quarter |
2.76 | 1.55 | ||||||
Fourth Quarter |
2.43 | 1.55 | ||||||
As of March 8, 2011, there were approximately 332 holders of record of our common stock.
The Company has not declared or paid any cash dividends on its common stock. No cash
dividends have been previously paid on our common stock and none are anticipated in fiscal 2011.
The Company also has shares of Series A and Series D Preferred Stock Outstanding that have
preferential dividend rights over any dividend payments to holders of common stock.
41
Table of Contents
Stock Performance Graph
COMPARISON OF 45 MONTH CUMULATIVE TOTAL RETURN*
Among Opko Health Inc, the NYSE Amex Composite Index
and the NYSE Arca Biotechnology Index
Among Opko Health Inc, the NYSE Amex Composite Index
and the NYSE Arca Biotechnology Index
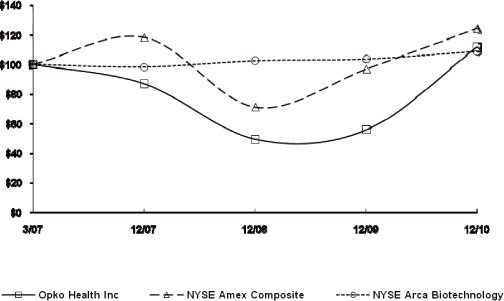
| *$100 invested on 3/27/07 in stock or 2/28/07 index, including reinvestment of dividends. Fiscal year ending December 31. |
||
42
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA.
The following selected historical consolidated statement of operations data for the years
ended December 31, 2010, 2009, 2008, and 2007 and for the period from inception (June 23, 2006)
through December 31, 2006 and the consolidated balance sheet data as of December 31, 2010, 2009,
2008, 2007, and 2006, below are derived from our audited consolidated financial statements and
related notes thereto. This data should be read in conjunction with our Managements Discussion
and Analysis of Financial Condition and Results of Operation and our consolidated financial
statements and the related notes thereto.
| For the period | ||||||||||||||||||||
| from inception | ||||||||||||||||||||
| June 23, 2006 | ||||||||||||||||||||
| through | ||||||||||||||||||||
| For the years ended December 31, | December 31, | |||||||||||||||||||
| (in thousands, except share and per shares information) | 2010 | 2009 | 2008 | 2007 | 2006 | |||||||||||||||
Statement of operations data |
||||||||||||||||||||
Revenue |
$ | 36,880 | $ | 13,147 | $ | 9,440 | $ | 847 | $ | | ||||||||||
Cost of goods sold |
20,501 | 9,567 | 8,559 | 808 | | |||||||||||||||
Gross margin |
16,379 | 3,580 | 881 | 39 | | |||||||||||||||
Operating expenses: |
||||||||||||||||||||
Selling, general and administrative |
22,121 | 13,518 | 14,790 | 12,466 | 375 | |||||||||||||||
Research and development |
7,908 | 12,881 | 21,562 | 10,850 | 508 | |||||||||||||||
Write-off of acquired in-process research and
development |
| 2,000 | 1,398 | 243,761 | | |||||||||||||||
Other operating expenses; primarily
amortization of
intangible assets |
3,579 | 3,201 | 1,694 | 150 | | |||||||||||||||
Total operating expenses |
33,608 | 31,600 | 39,444 | 267,227 | 883 | |||||||||||||||
Operating loss |
(17,229 | ) | (28,020 | ) | (38,563 | ) | (267,188 | ) | (883 | ) | ||||||||||
Other (expense) income, net |
(842 | ) | (2,034 | ) | (1,354 | ) | (671 | ) | 6 | |||||||||||
Loss before income taxes and investment losses |
(18,071 | ) | (30,054 | ) | (39,917 | ) | (267,859 | ) | (877 | ) | ||||||||||
Income tax
(expense) benefit |
(141 | ) | 294 | 83 | 83 | | ||||||||||||||
Net loss before investment losses |
(18,212 | ) | (29,760 | ) | (39,834 | ) | (267,776 | ) | (877 | ) | ||||||||||
Loss from investments in investees |
(714 | ) | (353 | ) | | (629 | ) | | ||||||||||||
Net loss |
(18,926 | ) | (30,113 | ) | (39,834 | ) | (268,405 | ) | (877 | ) | ||||||||||
Preferred stock dividend |
(2,624 | ) | (4,718 | ) | (217 | ) | (217 | ) | | |||||||||||
Net loss attributable to common shareholders |
$ | (21,550 | ) | $ | (34,831 | ) | $ | (40,051 | ) | $ | (268,622 | ) | $ | (877 | ) | |||||
Loss per share, basic and diluted |
$ | (0.08 | ) | $ | (0.15 | ) | $ | (0.21 | ) | $ | (2.09 | ) | $ | (0.01 | ) | |||||
Weighted average number of common shares
outstanding basic and diluted |
255,095,586 | 233,191,617 | 187,713,041 | 128,772,080 | 58,733,556 | |||||||||||||||
Balance sheet data |
||||||||||||||||||||
Total assets |
$ | 77,846 | $ | 87,430 | $ | 21,764 | $ | 39,568 | $ | 116 | ||||||||||
Working capital |
$ | 26,473 | $ | 50,795 | $ | 5,754 | $ | 19,489 | $ | 21 | ||||||||||
Long-term line of credit with related party, notes
payable, and capital lease obligations, net |
$ | | $ | 11,932 | $ | 11,867 | $ | 14,235 | $ | | ||||||||||
Series D Preferred Stock |
$ | 26,128 | $ | 26,128 | $ | | $ | | $ | | ||||||||||
Stockholders equity |
$ | 23,052 | $ | 31,599 | $ | 359 | $ | 16,784 | $ | 21 | ||||||||||
ITEM 7. MANAGEMENTS DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION.
This Annual Report on Form 10-K contains certain forward-looking statements within the meaning
of the Private Securities Litigation Reform Act of 1995 (PSLRA), Section 27A of the Securities
Act of 1933, as amended (the Securities Act), and Section 21E of the Securities Exchange Act of
1934, as amended, (the Exchange Act), about our expectations, beliefs, or intentions regarding
our product development efforts, business, financial condition, results of operations, strategies,
or prospects. You can identify forward-looking statements by the fact that these statements do not
relate strictly to historical or current matters. Rather, forward-looking statements relate to
anticipated or expected events, activities, trends, or results as of the date they are made.
Because forward-looking statements relate to matters that have not yet occurred, these statements
are inherently subject to risks and uncertainties that could cause our actual results to differ
materially from any future results expressed or implied by the forward-looking statements. Many
factors could cause our actual activities or results to differ materially from the activities and
results anticipated in forward-looking statements. These factors include those contained in Item
1A Risk Factors of this Annual Report on Form 10-K. We do not undertake any obligation to
update forward-looking statements. We intend that all forward-looking statements be subject to the
safe harbor provisions of PSLRA. These forward-looking statements are only predictions and reflect
our views as of the date they are made with respect to future events and financial performance.
43
Table of Contents
OVERVIEW
We are a multi-national pharmaceutical and diagnostics company that seeks to establish
industry-leading positions in large and rapidly growing medical markets by leveraging our
discovery, development and commercialization expertise and our novel and proprietary technologies.
Our current focus is on conditions with major unmet medical needs including neurological disorders,
infectious diseases, oncology and ophthalmologic diseases. We are developing a range of solutions
to diagnose, treat and prevent these conditions, including molecular diagnostics tests, proprietary
pharmaceuticals and vaccines. We plan to commercialize these solutions on a global basis in large
and high growth markets, including emerging markets. We have already established emerging markets
pharmaceutical platforms in Chile and Mexico, which are delivering revenue and which we expect to
deliver cash flow and facilitate future market entry for our products currently in development. We
also actively explore opportunities to acquire complementary pharmaceuticals, compounds,
technologies, and businesses.
We expect to incur substantial losses as we continue the development of our product
candidates, continue our other research and development activities, and establish a sales and
marketing infrastructure in anticipation of the commercialization of our diagnostic and
pharmaceutical product candidates. We currently have limited commercialization capabilities, and
it is possible that we may never successfully commercialize any of our diagnostic and
pharmaceutical product candidates. We do not currently generate revenue from any of our diagnostic
and pharmaceutical product candidates. Our research and development activities are budgeted to
expand over a period of time and will require further resources if we are to be successful. As a
result, we believe that our operating losses are likely to be substantial over the next several
years. We may need to obtain additional funds to further develop our research and development
programs, and there can be no assurance that additional capital will
be available to us when needed on
acceptable terms, or at all.
RECENT DEVELOPMENTS
On March 14, 2011, we issued 27,000,000 shares of our Common Stock in an underwritten public
offering at a price of $3.75 per share. The net proceeds received in connection with the offering
were approximately $96.4 million after deducting the underwriters discounts and commissions and
other estimated offering expenses. The offering closed on March 14, 2011. We also granted the
underwriters a 30-day option to purchase up to an additional 4,050,000 shares of our Common Stock
to cover overallotments, if any. On March 15, 2011, representatives for the underwriters provided us notice that the underwriters
exercised a portion of their 4,050,000 share over-allotment option for 2,397,029 additional shares of our Common
Stock.
On January 31, 2011, we acquired all of the outstanding stock of CURNA, Inc. (CURNA), a
privately held therapeutics company, in exchange for $10 million in cash. CURNA is engaged in the
discovery of new drugs for the treatment of a wide variety of illnesses, including cancer, heart
disease, metabolic disorders and a range of genetic anomalies.
RESULTS OF OPERATIONS
For The Years Ended December 31, 2010 and December 31, 2009
The results of operations for the year ended December 31, 2010 and 2009 include the post
acquisition operations for OPKO Chile (formerly known as Pharma Genexx, S.A.), a privately owned
Chilean company engaged in the marketing, sale, and distribution of pharmaceutical products,
devices, and over-the-counter products for government, private, and institutional markets in Chile,
which we acquired on October 7, 2009. As a result, our operating results for periods prior to
October 7, 2009 do not include any results related to OPKO Chile. Further, on February 16, 2010 we
acquired Exakta-OPKO (formerly known as Pharmacos Exakta, S.A. de C.V.), a privately owned Mexican
company engaged in the manufacture, marketing sale and distribution of pharmaceutical and
over-the-counter products for, private and institutional markets in Mexico. As such, our operating
results for periods prior to February 16, 2010 do not include any results related to Exakta-OPKO.
44
Table of Contents
Revenue.
Revenue for the year ended December 31, 2010 was $36.9 million compared to $13.1
million for the year ended December 31, 2009. Revenue from our pharmaceutical business for 2010
increased as compared to 2009 as a result of the revenue generated by our pharmaceutical
business through OPKO Chile and Exakta-OPKO and license revenue generated by the outlicense of our
NK-1 development program. In December 2010, we outlicensed our NK-1 development program to TESARO,
Inc. (TESARO) for an upfront cash payment of $6.0 million, future milestone payments of up to
$115.0 million, 1.5 million shares of TESARO Series O Preferred Stock (TESARO Preferred Stock),
as well as royalty payments on future sales. We recorded the TESARO Preferred Stock at fair value
and recognized $6.7 million as license revenue, including $6.0 million in cash and $0.7 million of
TESARO Preferred Stock.
Gross
margin. Gross margin for the year ended December 31, 2010 was $16.4 million compared to
$3.6 million for the year ended December 31, 2009. Gross margin improved during 2010 from gross
margin in 2009 as a result of our license revenue of $6.7 million related to TESARO, with no
associated cost of revenue, and increased gross margin generated by our pharmaceutical business
through OPKO Chile and Exakta-OPKO, partially offset by decreased margins from our instrumentation
business.
Selling, general and administrative expense. Selling, general and administrative expense in
the year ended December 31, 2010 was $22.1 million as compared to $13.5 million during the year
ended December 31, 2009. Selling, general and administrative expense increased primarily as a
result of expenses related to our pharmaceutical businesses in Chile and Mexico, as well as
increased personnel costs, including equity-based compensation, and professional fees. Included
in selling, general and administrative expenses were $5.1 million and $3.2 million of equity based
compensation expense for the years ended December 31, 2010 and 2009, respectively.
Research and development expense. Research and development expense for the year ended
December 31, 2010 was $7.9 million as compared to $12.9 million during the year ended December 31,
2009. Research and development expense decreased during 2010 primarily as a result of the 2009
period including activities related to our Phase III clinical trial for bevasiranib, which was
terminated in March 2009. Partially offsetting this decrease were increased activities related to
our rolapitant development program prior to its licensure to TESARO in December 2010 and increased
activities related to our molecular diagnostics program. In addition, during 2010 we received $0.7
million of grants under the New Qualifying Therapeutic Discovery Project Credit (or Grant) program
for expenditures related to certain development programs during 2009 and 2010. Further, we
received $0.3 million in research and development credits for certain development programs in
Mexico. Included in research and development expense were $1.7 million and $1.3 million of equity
based compensation expense for the years ended December 31, 2010 and 2009, respectively.
Write-off of acquired in-process research and development. On October 12, 2009, we entered
into an agreement to acquire certain assets from Schering Plough Corporations neurokinin-1
(NK-1) development program in an all cash transaction for $2.0 million at closing. We recorded
this acquisition as an asset acquisition and recorded the assets at fair value and allocated the
entire purchase price to acquired in-process research and development expense and recorded a charge
of $2.0 million.
We record expense for in-process research and development projects accounted for as asset
acquisitions which have not reached technological feasibility and which have no alternative future
use. The NK-1 drug candidates have not reached a stage of technological feasibility and have no
alternative future use. We did not have any in-process research and development activities during
2010.
Other operating expenses, principally amortization of intangible assets. Other operating
expenses were $3.6 million for the year ended December 31, 2010, compared to $3.2 million for the
year ended December 31, 2009. The increase in other operating expenses is a result of increased
intangible asset amortization related to our acquisitions of OPKO Chile and Exakta-OPKO. Partially
offsetting this increase, the 2009 period includes $1.1 million impairment of goodwill related to
our instrumentation business and there was no such impairment in 2010.
Other income and expenses. Other expense was $0.8 million for the year ended December 31,
2010, compared to $2.0 million for the year ended December 31, 2009. Other expenses primarily
consist of interest expense incurred on our $12.0 million line of credit with The Frost Group, LLC
(the Frost Group), a related party, partially offset by interest earned on our cash and cash
equivalents. The Frost Group members include the Frost Gamma Investment Trust (the Gamma Trust)
of which Phillip Frost, M.D., our Chairman and CEO, is the sole trustee, Jane Hsiao, the Companys
Vice Chairman and Chief Technical Officer, Steven D. Rubin, the Companys Executive Vice President
Administration and a director, and Rao Uppaluri, the Companys Chief Financial Officer.
45
Table of Contents
On June 2, 2010, we repaid all amounts outstanding on the line of credit including $12.0 million
in principal and $4.1 million in interest.
For The Years Ended December 31, 2009 and December 31, 2008
The results of operations for the year ended December 31, 2009 include the post acquisition
operations for OPKO Chile, which we acquired on October 7, 2009. As a result, our operating
results for periods prior to October 7, 2009 do not include OPKO Chile activity.
Revenue. Revenue for the year ended December 31, 2009 was $13.1 million compared to $9.4
million for the year ended December 31, 2008. Revenue for 2009 increased as a result of the
revenue generated by OPKO Chile after our acquisition on October 7, 2009. Partially offsetting the
revenue from OPKO Chile was decreased revenue from our instrumentation business in international
markets. Revenue for 2008 primarily consisted of revenue from our instrumentation business in
international markets.
Gross margin. Gross margin for the year ended December 31, 2009 was $3.6 million compared to
$0.9 million for the year ended December 31, 2008. Gross margin improved during 2009 primarily as
a result of improved gross margin from our instrumentation business and post acquisition gross
margin generated by our pharmaceutical business in Chile. Gross margin during 2008 was negatively
impacted while we made a number of changes to our instrumentation manufacturing process in an
effort to lower the cost of goods sold and increase gross margins. Those improvements included
changing suppliers of components for our OCT SLO products and assembling a number of components
in-house rather than outsourcing those activities. We realized the benefits of those changes to
our manufacturing processes as increased gross margin during the year ended December 31, 2009.
Selling, general and administrative expense. Selling, general and administrative expense in
the year ended December 31, 2009 was $13.5 million as compared to $14.8 million during the year
ended December 31, 2008. Selling, general and administrative expense decreased primarily as a
result of decreased personnel costs, including equity-based compensation, and sales commissions to
certain international distributors in our instrumentation business, partially offset by increased
professional fees. Included in selling, general and administrative expenses were $3.2 million and
$4.2 million of equity based compensation expense for the years ended December 31, 2009 and 2008,
respectively
Research and development expense. Research and development expense for the year ended
December 31, 2009 was $12.9 million as compared to $21.6 million during the year ended December 31,
2008. The decrease in research and development expense was primarily related to the termination of
the Phase III clinical trial of bevasiranib and related reduced personnel costs. Research and
development expense for the year ended December 31, 2009 consisted primarily of expenses related to
the Phase III clinical trial of bevasiranib through March 6, 2009 and the related costs of
analyzing the data generated by the trial. Research and development expense for the year ended
December 31, 2008 primarily consisted of expenses related to the Phase III clinical trial of
bevasiranib. Included in research and development expense were $1.3 million and $2.5 million of
equity based compensation expense for the years ended December 31, 2009 and 2008, respectively.
Write-off of acquired in-process research and development. On October 12, 2009, we entered
into an agreement to acquire certain assets from Schering Plough Corporations neurokinin-1
(NK-1) development program, of which, rolapitant was our lead pharmaceutical product candidate,
in an all cash transaction for $2.0 million at closing. We recorded this acquisition as an asset
acquisition and recorded the assets at fair value and allocated the entire purchase price to
acquired in-process research and development expense and recorded a charge of $2.0 million. On May
6, 2008, we acquired Vidus Ocular, Inc. in a stock for stock transaction. We recorded Vidus
assets and liabilities at fair value, and as a result, we recorded acquired in-process research and
development expense and recorded a charge of $1.4 million. We valued our common stock issued to
Vidus shareholders at the average closing price of the common stock on the date of the transaction
and two days prior to the transaction.
We record expense for in-process research and development projects accounted for as asset
acquisitions which have not reached technological feasibility and which have no alternative future
use. The NK-1 drug candidates have not reached a stage of technological feasibility and have no
alternative future use. At the time of our acquisition of Vidus, the accounting for business
combinations and asset acquisitions were the same and Vidus projects had not reached a stage of
technological feasibility and had no alternative future use. Effective January 1, 2009, in-process
research and development projects acquired in business combinations are capitalized.
46
Table of Contents
Other operating expenses, principally amortization of intangible assets. Other operating
expenses were $3.2 million for the year ended December 31, 2009, compared to $1.7 million for the
year ended December 31, 2008. The increase is primarily the result of the $1.1 million impairment
of goodwill related to our instrumentation business. In addition, the increase reflects the
amortization expense related to the intangible assets acquired as part of our acquisition of Pharma
Genexx.
Other income and expenses. Other expense was $2.0 million for the year ended December 31,
2009, compared to $1.4 million, net of $0.3 million of interest income for the year ended December
31, 2008. Other expenses primarily consist of interest expense incurred on our $12.0 million line
of credit, partially offset by interest earned on our cash and cash equivalents.
Liquidity And Capital Resources
At December 31, 2010, we had cash and cash equivalents of approximately $18.0 million compared
to $42.7 million on December 31, 2009. Cash used in operations during 2010 primarily reflects
expenses related to selling, general and administrative activities related to our corporate and
instrumentation operations, as well as our operations in Chile and Mexico. Partially offsetting
this, we received $6.0 million in cash from our outlicense to TESARO of our NK-1 development
program. Since our inception, we have not generated sufficient gross margins to offset our
operating and other expenses and our primary source of cash has been from the private placement of
stock and credit facilities available to us.
On March 14, 2011, we issued 27,000,000 shares of our Common Stock in an underwritten public
offering at a price of $3.75 per share. The net proceeds received in connection with the offering
were approximately $96.4 million after deducting the underwriters discounts and commissions and
other estimated offering expenses. We also granted the underwriters a 30-day option to purchase
up to an additional 4,050,000 shares of our Common Stock to cover overallotments, if any.
On January 31, 2011, we acquired all of the outstanding stock of CURNA, Inc. (CURNA), a
privately held therapeutics company, in exchange for $10.0 million in cash. CURNA is engaged in
the discovery of new drugs for the treatment of a wide variety of illnesses, including cancer,
heart disease, metabolic disorders and a range of genetic anomalies.
In connection with our acquisition of OPKO Chile, we have outstanding lines of credit in the
aggregate amount of $18.9 million with seven financial institutions in Chile, of which, $4.2
million is unused. The average interest rate on these lines of credit is approximately 6%. These
lines of credit are short-term and are generally due within three months. These lines of credit
are used primarily as a source of working capital for inventory purchases. The highest balance at
any time during the year ended December 31, 2010 was $14.8 million. We intend to continue to enter
into these lines of credit as needed. There is no assurance that this or other funding sources
will be available to us on acceptable terms, or at all.
We currently have an unutilized $12.0 million line of credit with the Frost Group. On June 2,
2010, we repaid all amounts outstanding on the line of credit including $12.0 million in principal
and $4.1 million in interest. The line of credit, which previously expired on January 11, 2011,
was renewed on February 22, 2011 on substantially the same terms as in effect at the time of
expiration. We are obligated to pay interest upon maturity, capitalized quarterly, on outstanding
borrowings under the line of credit at an 11% annual rate, which is due March 31, 2012. The line
of credit is collateralized by all of our U.S. based personal property except our intellectual
property.
We expect to incur losses from operations for the foreseeable future. We expect to incur
substantial research and development expenses, including expenses related to the hiring of
personnel and additional clinical trials. We expect that selling, general and administrative
expenses will also increase as we expand our sales, marketing and administrative staff and add
infrastructure.
We believe the cash and cash equivalents on hand at December 31, 2010, the amounts available
to be borrowed under our lines of credit, and proceeds from the issuance of our Common Stock in an
underwritten public offering on March 14, 2011, are sufficient to meet our anticipated cash
requirements for operations and debt service beyond the next 12 months. We based this estimate on
assumptions that may prove to be wrong or are subject to change, and we may be required to use our
available cash resources sooner than we currently expect. If we acquire additional assets or
companies, accelerate our product development programs or initiate additional clinical trials, we
will need additional funds. Our future cash requirements will depend on a number of factors,
including possible acquisitions, the continued progress of research and development of our product
candidates, the timing and outcome
47
Table of Contents
of clinical trials and regulatory approvals, the costs involved in preparing, filing,
prosecuting, maintaining, defending, and enforcing patent claims and other intellectual property
rights, the status of competitive products, the availability of financing, and our success in
developing markets for our product candidates. If we are not able to secure additional funding
when needed, we may have to delay, reduce the scope of, or eliminate one or more of our clinical
trials or research and development programs.
The following table provides information as of December 31, 2010 with respect to the amounts
and timing of our known contractual obligation payments due by period.
| Contractual obligations | After | |||||||||||||||||||||||||||
| (in thousands) | 2011 | 2012 | 2013 | 2014 | 2015 | 2015 | Total | |||||||||||||||||||||
Open purchase orders |
$ | 2,792 | $ | | $ | | $ | | $ | | $ | | $ | 2,792 | ||||||||||||||
Operating leases |
506 | 270 | | | | | 776 | |||||||||||||||||||||
Credit lines |
14,690 | | | | | | 14,690 | |||||||||||||||||||||
Total |
$ | 17,988 | $ | 270 | $ | | $ | | $ | | $ | | $ | 18,258 | ||||||||||||||
The preceding table does not include information where the amounts of the obligations are not
currently determinable, including contractual obligations in connection with product license
agreements that include payments upon achievement of certain milestones.
Critical Accounting Policies and Estimates
Accounting Estimates. The preparation of financial statements in conformity with generally
accepted accounting principles requires management to make estimates and assumptions that affect
the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities
at the date of the financial statements and the reported amounts of sales and expenses during the
reporting period. Actual results could differ from those estimates.
Equity-based compensation. We recognize equity based compensation as an expense in our
financial statements and that cost is measured at the fair value of the award and expensed over
their vesting period. Equity-based compensation arrangements to non-employees are recorded at
their fair value on the measurement date. We estimate the grant-date fair value of our stock
option grants using a valuation model known as the Black-Scholes-Merton formula or the
Black-Scholes Model and allocate the resulting compensation expense over the corresponding
requisite service period associated with each grant. The Black-Scholes Model requires the use of
several variables to estimate the grant-date fair value of stock options including expected term,
expected volatility, expected dividends and risk-free interest rate. We perform significant
analyses to calculate and select the appropriate variable assumptions used in the Black-Scholes
Model. We also perform significant analyses to estimate forfeitures of equity-based awards. We
are required to adjust our forfeiture estimates on at least an annual basis based on the number of
share-based awards that ultimately vest. The selection of assumptions and estimated forfeiture
rates is subject to significant judgment and future changes to our assumptions and estimates may
have a material impact on our Consolidated Financial Statements.
Goodwill and intangible assets. The allocation of the purchase price for acquisitions
requires extensive use of accounting estimates and judgments to allocate the purchase price to the
identifiable tangible and intangible assets acquired, including in-process research and
development, and liabilities assumed based on their respective fair values. Additionally, we must
determine whether an acquired entity is considered to be a business or a set of net assets, because
a portion of the purchase price can only be allocated to goodwill in a business combination.
Appraisals inherently require significant estimates and assumptions, including but not limited
to, determining the timing and estimated costs to complete the in-process research and development
projects, projecting regulatory approvals, estimating future cash flows, and developing appropriate
discount rates. We believe the estimated fair values assigned to the Exakta-OPKO assets acquired
and liabilities assumed are based on reasonable assumptions. However, the fair value estimates for
the purchase price allocation may change during the allowable allocation period, which is up to one
year from the acquisition date, if additional information becomes available that would require
changes to our estimates.
Allowance for doubtful accounts and revenue recognition. Generally, we recognize revenue from
product sales when goods are shipped and title and risk of loss transfer to our customers. Certain
of our products are sold directly to end-users and require that we deliver, install and train the
staff at the end-users facility. As a result, we do not recognize revenue until the product is
delivered, installed and training has occurred. Return policies in certain international markets
for our medical device products provide for stringent guidelines in accordance with the terms
48
Table of Contents
of contractual agreements with customers. Our estimates for sales returns are based upon the
historical patterns of products returned matched against the sales from which they originated, and
managements evaluation of specific factors that may increase the risk of product returns. We
analyze accounts receivable and historical bad debt levels, customer credit worthiness and current
economic trends when evaluating the adequacy of the allowance for doubtful accounts using the
specific identification method. Our reported net loss is directly affected by managements
estimate of the collectability of accounts receivable. The allowance for doubtful accounts
recognized in our consolidated balance sheets at December 31, 2010 and December 31, 2009 was $1.2
million and $0.4 million, respectively.
Recent accounting pronouncements. In December 2010, the FASB issued an amendment to the
disclosure of supplementary pro forma information for business combinations. The amendment
specifies that if a public entity presents comparative financial statements, the entity should
disclose revenue and earnings of the combined entity as though the business combination(s) that
occurred during the current year had occurred as of the beginning of the comparable prior annual
reporting period only. The amendment also expands the supplemental pro forma disclosures under
current accounting guidance to include a description of the nature and amount of material,
nonrecurring pro forma adjustments directly attributable to the business combination included in
the reported pro forma revenue and earnings. The amendment is effective prospectively for business
combinations for which the acquisition date is on or after the beginning of the first annual
reporting period beginning on or after December 15, 2010. Early adoption is permitted. The
adoption of this amendment is not expected to have a material impact on our financial statement
disclosures.
In December 2010, the FASB issued an amendment to the accounting for goodwill impairment
tests. The amendment modifies Step 1 of the impairment test for reporting units with zero or
negative carrying amounts. For those reporting units, an entity is required to perform Step 2 of
the goodwill impairment test if it is more likely than not that a goodwill impairment exists. In
determining whether it is more likely than not that a goodwill impairment exists, an entity should
consider whether there are any adverse qualitative factors indicating that an impairment may exist.
The qualitative factors are consistent with the existing guidance. The amendment is effective for
fiscal years, and interim periods within those years, beginning after December 15, 2010. Early
adoption is not permitted. The adoption of this amendment is not expected to have a material
impact on our results of operations or financial condition.
In December 2010, the FASB issued an amendment to the accounting for annual excise taxes paid
to the federal government by pharmaceutical manufacturers under health care reform. The liability
for the fee should be estimated and recorded in full upon the first qualifying branded prescription
drug sale with a corresponding deferred cost that is amortized to expense using a straight-line
method of allocation unless another method better allocates the fee over the calendar year that it
is payable. The amendment is effective for calendar years beginning after December 31, 2010, when
the fee initially becomes effective. As we currently do not
manufacture pharmaceutical products, we do not expect the adoption of
this amendment to have a material impact on our results of operations or financial condition.
In March 2010, the FASB reached a consensus to issue an amendment to the accounting for
revenue arrangements under which a vendor satisfies its performance obligations to a customer over
a period of time, when the deliverable or unit of accounting is not within the scope of other
authoritative literature, and when the arrangement consideration is contingent upon the achievement
of a milestone. The amendment defines a milestone and clarifies whether an entity may recognize
consideration earned from the achievement of a milestone in the period in which the milestone is
achieved. This amendment is effective for fiscal years beginning on or after June 15, 2010, with
early adoption permitted. The amendment may be applied retrospectively to all arrangements or
prospectively for milestones achieved after the effective date. We have not adopted this guidance
early and adoption of this amendment is not expected to have a material impact on our results of
operation or financial condition.
49
Table of Contents
In January 2010, the FASB issued an amendment to the accounting for fair value measurements
and disclosures. This amendment details additional disclosures on fair value measurements,
requires a gross presentation of activities within a Level 3 rollforward, and adds a new
requirement to disclose transfers in and out of Level 1 and Level 2 measurements. The new
disclosures are required of all entities that are required to provide disclosures about recurring
and nonrecurring fair value measurements. This amendment is effective in the first interim or
reporting period beginning after December 15, 2009, with an exception for the gross presentation of
Level 3 rollforward information, which is required for annual reporting periods beginning after
December 15, 2010, and for interim reporting periods within those years. The adoption of this
amendment did not have a material impact on our financial statement disclosures.
In October 2009, the FASB issued an amendment to the accounting for multiple-deliverable
revenue arrangements. This amendment provides guidance on whether multiple deliverables exist, how
the arrangements should be separated, and how the consideration paid should be allocated. As a
result of this amendment, entities may be able to separate multiple-deliverable arrangements in
more circumstances than under existing accounting guidance. This guidance amends the requirement
to establish the fair value of undelivered products and services based on objective evidence and
instead provides for separate revenue recognition based upon managements best estimate of the
selling price for an undelivered item when there is no other means to determine the fair value of
that undelivered item. The existing guidance previously required that the fair value of the
undelivered item reflect the price of the item either sold in a separate transaction between
unrelated third parties or the price charged for each item when the item is sold separately by the
vendor. If the fair value of all of the elements in the arrangement was not determinable, then
revenue was deferred until all of the items were delivered or fair value was determined. This
amendment will be effective prospectively for revenue arrangements entered into or materially
modified in fiscal years beginning on or after June 15, 2010. Early adoption and retrospective
application is also permitted. We are currently evaluating the potential effect of the adoption of
this amendment on our results of operations or financial condition.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
In the normal course of doing business, we are exposed to the risks associated with foreign
currency exchange rates and changes in interest rates.
Foreign Currency Exchange Rate Risk Although we do not speculate in the foreign exchange
market, we may from time to time manage exposures that arise in the normal course of business
related to fluctuations in foreign currency exchange rates by entering into offsetting positions
through the use of foreign exchange forward contracts. Certain firmly committed transactions are
hedged with foreign exchange forward contracts. As exchange rates change, gains and losses on the
exposed transactions are partially offset by gains and losses related to the hedging contracts.
Both the exposed transactions and the hedging contracts are translated at current spot rates, with
gains and losses included in earnings.
Our derivative activities, which consist of foreign exchange forward contracts, are initiated
to hedge forecasted cash flows that are exposed to foreign currency risk. The foreign exchange
forward contracts generally require us to exchange local currencies for foreign currencies based on
pre-established exchange rates at the contracts maturity dates. As exchange rates change, gains
and losses on these contracts are generated based on the change in the exchange rates that are
recognized in the consolidated statement of operations at maturity, and offset the impact of the
change in exchange rates on the foreign currency cash flows that are hedged. If the counterparties
to the exchange contracts do not fulfill their obligations to deliver the contracted currencies, we
could be at risk for currency related fluctuations. We enter into these contracts with
counterparties that we believe to be creditworthy
50
Table of Contents
and do not enter into any leveraged derivative transactions. We had $7.7 million in foreign
exchange forward contracts outstanding at December 31, 2010 and $6.3 million at December 31, 2009
primarily to hedge Chilean-based operating cash flows against US dollars. If Chilean Pesos were to
strengthen in relation to the US dollar, our loss or gain on hedged foreign currency cash-flows would be offset by
the derivative contracts, with a net effect of zero.
We do not engage in trading market risk sensitive instruments or purchasing hedging
instruments or other than trading instruments that are likely to expose us to significant market
risk, whether interest rate, foreign currency exchange, commodity price, or equity price risk.
Interest Rate Risk Our exposure to market risk relates to our cash and investments and to
our borrowings. We maintain an investment portfolio of money market funds. The securities in our
investment portfolio are not leveraged, and are, due to their very short-term nature, subject to
minimal interest rate risk. We currently do not hedge interest rate exposure. Because of the
short-term maturities of our investments, we do not believe that a change in market interest rates
would have a significant negative impact on the value of our investment portfolio except for
reduced income in a low interest rate environment. At December 31, 2010, we had cash and cash
equivalents of $18.0 million. The weighted average interest rate related to our cash and cash
equivalents for the year ended December 31, 2010 was 0%. As of December 31, 2010, the principal
value of our credit lines was $14.7 million, and have a weighted average interest rate of 6%.
The primary objective of our investment activities is to preserve principal while at the same
time maximizing yields without significantly increasing risk. To achieve this objective, we invest
our excess cash in debt instruments of the U.S. Government and its agencies, bank obligations,
repurchase agreements and high-quality corporate issuers, and money market funds that invest in
such debt instruments, and, by policy, restrict our exposure to any single corporate issuer by
imposing concentration limits. To minimize the exposure due to adverse shifts in interest rates,
we maintain investments at an average maturity of generally less than three months.
51
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
| Page | ||||
| 53 | ||||
| 55 | ||||
| 56 | ||||
| 57 | ||||
| 59 | ||||
| 60 - 84 |
52
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of OPKO Health, Inc. and subsidiaries
We have audited the accompanying consolidated balance sheets of OPKO Health, Inc. and
subsidiaries as of December 31, 2010 and 2009, and the related consolidated statements of
operations, shareholders equity, and cash flows for each of the three years in the period ended
December 31, 2010. These financial statements are the responsibility of the Companys management.
Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether the financial statements are free of material
misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and
disclosures in the financial statements. An audit also includes assessing the accounting
principles used and significant estimates made by management, as well as evaluating the overall
financial statement presentation. We believe that our audits provide a reasonable basis for our
opinion.
In our opinion, the financial statements referred to above present fairly, in all material
respects, the consolidated financial position of OPKO Health, Inc. and subsidiaries at December 31,
2010 and 2009, and the consolidated results of their operations and their cash flows for each of
the three years in the period ended December 31, 2010, in conformity with U.S. generally accepted
accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), OPKO Health Inc. and subsidiaries internal control over financial
reporting as of December 31, 2010, based on criteria established in Internal Control-Integrated
Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our
report dated March 16, 2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Certified Public Accountants
Certified Public Accountants
Miami, Florida
March 16, 2011
March 16, 2011
53
Table of Contents
Report of Independent Registered Public Accounting Firm
The Board of Directors and Shareholders of OPKO Health, Inc. and subsidiaries
We have audited OPKO Health, Inc. and subsidiaries internal control over financial reporting
as of December 31, 2010, based on criteria established in Internal ControlIntegrated Framework
issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria).
OPKO Health, Inc. and subsidiaries management is responsible for maintaining effective internal
control over financial reporting, and for its assessment of the effectiveness of internal control
over financial reporting included in the accompanying Managements Annual Report on Internal
Control Over Financial Reporting. Our responsibility is to express an opinion on the companys
internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting
Oversight Board (United States). Those standards require that we plan and perform the audit to
obtain reasonable assurance about whether effective internal control over financial reporting was
maintained in all material respects. Our audit included obtaining an understanding of internal
control over financial reporting, assessing the risk that a material weakness exists, testing and
evaluating the design and operating effectiveness of internal control based on the assessed risk,
and performing such other procedures as we considered necessary in the circumstances. We believe
that our audit provides a reasonable basis for our opinion.
A companys internal control over financial reporting is a process designed to provide
reasonable assurance regarding the reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with generally accepted accounting
principles. A companys internal control over financial reporting includes those policies and
procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately
and fairly reflect the transactions and dispositions of the assets of the company; (2) provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting principles, and that receipts and
expenditures of the company are being made only in accordance with authorizations of management and
directors of the company; and (3) provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use or disposition of the companys assets that could have a
material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent
or detect misstatements. Also, projections of any evaluation of effectiveness to future periods
are subject to the risk that controls may become inadequate because of changes in conditions or
that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, OPKO Health, Inc. and subsidiaries maintained, in all material respects,
effective internal control over financial reporting as of December 31, 2010, based on the COSO
criteria.
We also have audited, in accordance with the standards of the Public Company Accounting
Oversight Board (United States), the consolidated balance sheets of OPKO Health, Inc. and
subsidiaries as of December 31, 2010 and 2009, and the related consolidated statements of
operations, shareholders equity, and cash flows for each of the three years in the period ended
December 31, 2010 of OPKO Health, Inc. and subsidiaries, and our
report dated March 16, 2011
expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Certified Public Accountants
Certified Public Accountants
Miami, Florida
March 16, 2011
March 16, 2011
54
Table of Contents
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED BALANCE SHEETS
(in thousands, except share data)
| December 31, | ||||||||
| 2010 | 2009 | |||||||
ASSETS |
||||||||
Current assets |
||||||||
Cash and cash equivalents |
$ | 18,016 | $ | 42,658 | ||||
Accounts receivable, net |
13,317 | 8,767 | ||||||
Inventory, net |
19,957 | 10,520 | ||||||
Prepaid expenses and other current assets |
2,782 | 1,873 | ||||||
Total current assets |
54,072 | 63,818 | ||||||
Property and equipment, net |
2,729 | 593 | ||||||
Intangible assets, net |
9,964 | 12,722 | ||||||
Goodwill |
5,856 | 5,408 | ||||||
Investments, net |
5,114 | 4,447 | ||||||
Other assets |
111 | 442 | ||||||
Total assets |
$ | 77,846 | $ | 87,430 | ||||
LIABILITIES, SERIES D PREFERRED STOCK AND SHAREHOLDERS EQUITY |
||||||||
Current liabilities |
||||||||
Accounts payable |
$ | 7,170 | $ | 4,784 | ||||
Accrued expenses |
5,739 | 3,918 | ||||||
Current portion of lines of credit and notes payable |
14,690 | 4,321 | ||||||
Total current liabilities |
27,599 | 13,023 | ||||||
Long-term liabilities interest payable to related party |
| 3,409 | ||||||
Other long-term liabilities, principally deferred tax liabilities |
1,067 | 1,339 | ||||||
Line of credit with related party, net of unamortized discount of $0 and
$68, respectively |
| 11,932 | ||||||
Total liabilities |
28,666 | 29,703 | ||||||
Commitments and contingencies |
||||||||
Series D Preferred Stock $0.01 par value, 2,000,000 shares authorized;
1,209,677 and 1,209,677 shares issued and outstanding (liquidation value
of $33,013 and $30,613) at December 31, 2010 and 2009, respectively |
26,128 | 26,128 | ||||||
Shareholders equity |
||||||||
Series A Preferred Stock $0.01 par value, 4,000,000 shares authorized;
897,439 and 1,025,934 shares issued and outstanding (liquidation value
of $2,468 and $2,564) at December 31, 2010 and 2009, respectively |
9 | 10 | ||||||
Series C Preferred Stock $0.01 par value, 500,000 shares authorized;
No shares issued or outstanding |
| | ||||||
Common Stock $0.01 par value, 500,000,000 shares authorized;
255,412,706 and 253,762,552 shares issued and outstanding at
December 31, 2010 and 2009, respectively |
2,554 | 2,538 | ||||||
Treasury stock (45,154 shares at December 31, 2010
and 2009) |
(61 | ) | (61 | ) | ||||
Additional paid-in capital |
376,008 | 367,028 | ||||||
Accumulated other comprehensive income |
2,921 | 1,313 | ||||||
Accumulated deficit |
(358,379 | ) | (339,229 | ) | ||||
Total shareholders equity |
23,052 | 31,599 | ||||||
Total
liabilities, Series D Preferred Stock and shareholders equity |
$ | 77,846 | $ | 87,430 | ||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these
statements.
55
Table of Contents
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except share data)
| For the years ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
Revenue |
||||||||||||
Products |
$ | 30,149 | $ | 13,147 | $ | 9,440 | ||||||
License |
6,731 | | | |||||||||
Total revenue |
36,880 | 13,147 | 9,440 | |||||||||
Cost of goods sold, excluding amortization
of intangible assets |
20,501 | 9,567 | 8,559 | |||||||||
Gross margin, excluding amortization of
intangible assets |
16,379 | 3,580 | 881 | |||||||||
Operating expenses |
||||||||||||
Selling, general and administrative |
22,121 | 13,518 | 14,790 | |||||||||
Research and development |
7,908 | 12,881 | 21,562 | |||||||||
Write-off of acquired in-process research and
development |
| 2,000 | 1,398 | |||||||||
Other operating expenses, principally
amortization of intangible assets |
3,579 | 3,201 | 1,694 | |||||||||
Total operating expenses |
33,608 | 31,600 | 39,444 | |||||||||
Operating loss |
(17,229 | ) | (28,020 | ) | (38,563 | ) | ||||||
Other expense, net |
(842 | ) | (2,034 | ) | (1,354 | ) | ||||||
Loss before income taxes and investment losses |
(18,071 | ) | (30,054 | ) | (39,917 | ) | ||||||
Income tax (expense) benefit |
(141 | ) | 294 | 83 | ||||||||
Loss before investment losses |
(18,212 | ) | (29,760 | ) | (39,834 | ) | ||||||
Loss from investments in investees |
(714 | ) | (353 | ) | | |||||||
Net loss |
(18,926 | ) | (30,113 | ) | (39,834 | ) | ||||||
Preferred stock dividend |
(2,624 | ) | (4,718 | ) | (217 | ) | ||||||
Net loss attributable to common shareholders |
$ | (21,550 | ) | $ | (34,831 | ) | $ | (40,051 | ) | |||
Loss per share, basic and diluted |
$ | (0.08 | ) | $ | (0.15 | ) | $ | (0.21 | ) | |||
Weighted average number of common shares
outstanding, basic and diluted |
255,095,586 | 233,191,617 | 187,713,041 | |||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these
statements.
56
Table of Contents
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF SHAREHOLDERS EQUITY
(in thousands, except share data)
For the years ended December 31, 2008, 2009 and 2010
| Series A Preferred | Additional | |||||||||||||||||||||||||||||||||||||||||||||||
| Stock | Series C Preferred Stock | Common Stock | Treasury | Paid-In | Other Comprehensive | Accumulated | ||||||||||||||||||||||||||||||||||||||||||
| Shares | Dollars | Shares | Dollars | Shares | Dollars | Shares | Dollars | Capital | Income | Deficit | Total | |||||||||||||||||||||||||||||||||||||
Balance at December 31, 2007 |
954,799 | $ | 10 | | $ | | 178,344,608 | $ | 1,783 | | | $ | 284,273 | $ | | $ | (269,282 | ) | $ | 16,784 | ||||||||||||||||||||||||||||
Equity-based compensation expense |
| | | | | | | | 6,730 | | | 6,730 | ||||||||||||||||||||||||||||||||||||
Issuance of equity securities to
acquire Vidus Ocular, Inc. at
$1.65 per share |
| | | | 658,080 | 7 | | | 1,312 | | | 1,319 | ||||||||||||||||||||||||||||||||||||
Correction of equity securities to
Acuity |
| | | | 57,408 | 1 | | | (1 | ) | | | | |||||||||||||||||||||||||||||||||||
Exercise of common stock options |
| | | | 5,187,149 | 52 | | | 154 | | | 206 | ||||||||||||||||||||||||||||||||||||
Exercise of common warrants |
| | | | 1,171,899 | 12 | | | 165 | | | 177 | ||||||||||||||||||||||||||||||||||||
Issuance of common stock in private
placement to related party at 1.11 |
| | | | 13,513,514 | 135 | | | 14,865 | | | 15,000 | ||||||||||||||||||||||||||||||||||||
Series A preferred stock dividend |
86,678 | 1 | | | | | | | | | | 1 | ||||||||||||||||||||||||||||||||||||
Conversion of Series A preferred
stock |
(87,721 | ) | (1 | ) | | | 87,721 | 1 | | | | | | | ||||||||||||||||||||||||||||||||||
Other disposition of assets for stock |
| | | | | | (18,000 | ) | (24 | ) | | | | (24 | ) | |||||||||||||||||||||||||||||||||
Net loss for the year ended
December 31, 2008 |
| | | | | | | | | | (39,834 | ) | (39,834 | ) | ||||||||||||||||||||||||||||||||||
Balance at December 31, 2008 |
953,756 | 10 | | | 199,020,379 | 1,991 | (18,000 | ) | (24 | ) | 307,498 | | (309,116 | ) | 359 | |||||||||||||||||||||||||||||||||
Equity-based compensation expense |
| | | | | | | | 4,498 | | | 4,498 | ||||||||||||||||||||||||||||||||||||
Exercise of common stock options |
| | | | 2,984,945 | 30 | | | 664 | | | 694 | ||||||||||||||||||||||||||||||||||||
Exercise of common warrants |
| | | | 706,164 | 7 | | | 17 | | | 24 | ||||||||||||||||||||||||||||||||||||
Issuance of common stock in
private placement with related
parties at $1.00 per share |
| | | | 20,000,000 | 200 | | | 19,800 | | | 20,000 | ||||||||||||||||||||||||||||||||||||
Issuance of common stock in private
placement, including related
parties, at $1.00 per share |
| | | | 31,000,000 | 310 | | | 30,680 | | | 30,990 | ||||||||||||||||||||||||||||||||||||
Relative fair value of warrants issued
In connection with issuance of 8%
Series D preferred stock |
| | | | | | | | 3,872 | | | 3,872 | ||||||||||||||||||||||||||||||||||||
Series A preferred stock dividend |
93,242 | 1 | | | | | | | (1 | ) | | | | |||||||||||||||||||||||||||||||||||
Conversion of Series A preferred
stock |
(21,064 | ) | (1 | ) | | | 21,064 | | | | | | | (1 | ) | |||||||||||||||||||||||||||||||||
Restricted stock grant |
| | | | 30,000 | | | | | | | | ||||||||||||||||||||||||||||||||||||
Purchase of shares at $3.55 |
| | | | | | (27,154 | ) | (37 | ) | | | | (37 | ) | |||||||||||||||||||||||||||||||||
Net loss for the year ended
December 31, 2009 |
| | | | | | | | | | (30,113 | ) | (30,113 | ) | ||||||||||||||||||||||||||||||||||
Cumulative
translation adjustment net |
| | | | | | | | | 1,313 | | 1,313 | ||||||||||||||||||||||||||||||||||||
Other
comprehensive loss |
(28,800 | ) | ||||||||||||||||||||||||||||||||||||||||||||||
Balance at December 31, 2009 |
1,025,934 | 10 | | | 253,762,552 | 2,538 | (45,154 | ) | (61 | ) | 367,028 | 1,313 | (339,229 | ) | 31,599 | |||||||||||||||||||||||||||||||||
57
Table of Contents
| Series A Preferred | Additional | |||||||||||||||||||||||||||||||||||||||||||||||
| Stock | Series C Preferred Stock | Common Stock | Treasury | Paid-In | Other Comprehensive | Accumulated | ||||||||||||||||||||||||||||||||||||||||||
| Shares | Dollars | Shares | Dollars | Shares | Dollars | Shares | Dollars | Capital | Income | Deficit | Total | |||||||||||||||||||||||||||||||||||||
Equity-based compensation expense |
| | | | | | | | 6,922 | | | 6,922 | ||||||||||||||||||||||||||||||||||||
Exercise of common stock options |
| | | | 150,231 | 2 | | | 72 | | | 74 | ||||||||||||||||||||||||||||||||||||
Series A preferred stock dividend |
| | | | | | | | | | (224 | ) | (224 | ) | ||||||||||||||||||||||||||||||||||
Conversion of Series A preferred
stock |
(128,495 | ) | (1 | ) | | | 128,495 | 1 | | | | | | | ||||||||||||||||||||||||||||||||||
Issuance of common stock to
acquire Pharmacos Exakta
at $1.46 per share |
| | | | 1,371,428 | 13 | | | 1,986 | | | 1,999 | ||||||||||||||||||||||||||||||||||||
Net loss for the year ended
December 31, 2010 |
| | | | | | | | | | (18,926 | ) | (18,926 | ) | ||||||||||||||||||||||||||||||||||
Cumulative translation adjustment
net |
| | | | | | | | | 1,608 | | 1,608 | ||||||||||||||||||||||||||||||||||||
Other
comprehensive loss |
(17,318 | ) | ||||||||||||||||||||||||||||||||||||||||||||||
Balance at December 31, 2010 |
897,439 | $ | 9 | | $ | | 255,412,706 | $ | 2,554 | (45,154 | ) | $ | (61 | ) | $ | 376,008 | $ | 2,921 | $ | (358,379 | ) | $ | 23,052 | |||||||||||||||||||||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these
statements.
58
Table of Contents
OPKO Health, Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| For the years ended | ||||||||||||
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
Cash flows from operating activities: |
||||||||||||
Net loss |
$ | (18,926 | ) | $ | (30,113 | ) | $ | (39,834 | ) | |||
Adjustments to reconcile net loss to net cash used in
operating activities: |
||||||||||||
Depreciation and amortization |
3,870 | 2,357 | 1,823 | |||||||||
Impairment of goodwill |
| 1,097 | | |||||||||
Write-off of acquired in-process research and development |
| 2,000 | 1,398 | |||||||||
Accretion of debt discount related to notes payable |
66 | 123 | 190 | |||||||||
Losses from investments in investees |
714 | 353 | | |||||||||
Equity based compensation employees and non-employees |
6,922 | 4,498 | 6,730 | |||||||||
Provision for bad debts |
446 | 73 | 204 | |||||||||
Provision for inventory obsolescence |
64 | 279 | 255 | |||||||||
Foreign exchange |
(382 | ) | 122 | | ||||||||
Loss on disposal of assets |
| | 148 | |||||||||
License of product for equity |
(731 | ) | | | ||||||||
Changes in: |
||||||||||||
Accounts receivable |
(3,396 | ) | (1,271 | ) | 590 | |||||||
Inventory |
(7,589 | ) | (928 | ) | (2,104 | ) | ||||||
Prepaid expenses and other current assets |
894 | 431 | 25 | |||||||||
Other assets |
420 | (276 | ) | | ||||||||
Accounts payable |
1,756 | (1,019 | ) | (1,225 | ) | |||||||
Accrued expenses |
(3,222 | ) | (1,062 | ) | 2,506 | |||||||
Net cash used in operating activities |
(19,094 | ) | (23,336 | ) | (29,294 | ) | ||||||
Cash flows from investing activities: |
||||||||||||
Investments in investees |
(650 | ) | (4,800 | ) | | |||||||
Acquisition of businesses, net of cash |
(1,323 | ) | (15,632 | ) | 48 | |||||||
Acquisition of rolapitant |
| (2,000 | ) | | ||||||||
Purchase of marketable securities |
(14,997 | ) | (9,997 | ) | | |||||||
Maturities of marketable securities |
14,997 | 9,997 | | |||||||||
Capital expenditures |
(807 | ) | (172 | ) | (378 | ) | ||||||
Net cash used in investing activities |
(2,780 | ) | (22,604 | ) | (330 | ) | ||||||
Cash flows from financing activities: |
||||||||||||
Issuance of common stock for cash to related party |
| 30,990 | 15,000 | |||||||||
Issuance of common stock |
| 20,000 | | |||||||||
Issuance of Series D preferred stock and warrants,
including related parties |
| 30,000 | | |||||||||
Repayments of line of credit with related party |
(12,000 | ) | | | ||||||||
Proceeds from bridge loan with related party |
| 3,000 | | |||||||||
Repayment of bridge loan with related party |
| (3,000 | ) | | ||||||||
Insurance financing and borrowings on lines of credit |
15,424 | 529 | 371 | |||||||||
Proceeds from the exercise of stock options and warrants |
74 | 718 | 383 | |||||||||
Repayments of notes payable and capital lease obligations |
(6,266 | ) | (317 | ) | (2,825 | ) | ||||||
Net cash (used in) provided by financing activities |
(2,768 | ) | 81,920 | 12,929 | ||||||||
Net change in cash and cash equivalents |
(24,642 | ) | 35,980 | (16,695 | ) | |||||||
Cash and cash equivalents at beginning of year |
42,658 | 6,678 | 23,373 | |||||||||
Cash and cash equivalents at end of year |
$ | 18,016 | $ | 42,658 | $ | 6,678 | ||||||
The accompanying Notes to Consolidated Financial Statements are an integral part of these
statements.
59
Table of Contents
OPKO Health, Inc. and Subsidiaries
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 Business and Organization
We are a multi-national pharmaceutical and diagnostics company that seeks to establish
industry-leading positions in large and rapidly growing medical markets by leveraging our
discovery, development and commercialization expertise and our novel and proprietary technologies.
Our current focus is on conditions with major unmet medical needs including neurological disorders,
infectious diseases, oncology and ophthalmologic diseases. We are developing a range of solutions
to diagnose, treat and prevent these conditions, including molecular diagnostics tests, proprietary
pharmaceuticals and vaccines. We plan to commercialize these solutions on a global basis in large
and high growth markets, including emerging markets. We have already established emerging markets
pharmaceutical platforms in Chile and Mexico, which are delivering revenue and which we expect to
deliver cash flow and facilitate future market entry for our products currently in development. We
also actively explore opportunities to acquire complementary pharmaceuticals, compounds,
technologies, and businesses. We are a Delaware corporation, headquartered in Miami, Florida.
Note 2 Acquisitions, Investments, and Licenses
Rolapitant license
In December 2010, we entered into a license agreement (the TESARO License) with TESARO, Inc.
(TESARO) granting TESARO exclusive rights to the development, manufacture, commercialization and
distribution of rolapitant and a related compound. Under the terms of the TESARO License, we are
eligible for payments of up to $121.0 million, including an up-front payment of $6.0 million, which
has been received, and additional payments based upon achievement of specified regulatory and
commercialization milestones. In addition, TESARO will pay us double digit tiered royalties on
sales of licensed product. We will share future profits from the commercialization of licensed
products in Japan with TESARO and we will have an option to market the products in Latin America.
In connection with the TESARO License, we also acquired a 10% equity position in TESARO. We
recorded the 10% equity position at $0.7 million, the estimated fair value based on a discounted
cash flow model.
In accounting for the license of rolapitant to TESARO, we determined that we did not have any
continuing involvement in the development of rolapitant or any other future performance obligations
and, as a result, recognized the $6.0 million up-front payment and the $0.7 million equity position
as license revenue during the year ended December 31, 2010.
We acquired rolapitant on October 12, 2009 from Schering-Plough Corporation (Schering). We
entered into an asset purchase agreement (the Schering Agreement) with Schering to acquire
rolapitant and other assets relating to Scherings neurokinin-1 (NK-1) receptor antagonist
program. Under the terms of the Schering Agreement, we paid Schering $2.0 million in cash upon
closing and agreed to pay up to an additional $27.0 million upon certain development milestones.
Rolapitant, the lead product in the NK-1 program, successfully completed Phase II clinical testing
for prevention of nausea and vomiting related to cancer chemotherapy and surgery, and other
indications. Development of rolapitant and the other assets had been stopped at the time of our
acquisition and there were no ongoing clinical trials. None of the assets acquired have
alternative future uses, nor have they reached a stage of technological feasibility, as such, we
recorded $2.0 million as in-process research and development expense during the year ended December
31, 2009.
Latin America acquisitions
In February 2010, we acquired Exakta-OPKO (previously known as Pharmacos Exakta S.A. de C.V.),
a privately-owned Mexican company, engaged in the manufacture, marketing and distribution of
ophthalmic and other pharmaceutical products for government and private markets since 1957.
Pursuant to a purchase agreement we acquired all of the outstanding stock of Exakta-OPKO and real
property owned by an affiliate of Exakta-OPKO for a total aggregate purchase price of $3.5 million,
of which an aggregate of $1.5 million was paid in cash and $2.0 million was paid in shares of our
Common Stock, par value $.01. In September 2010, we reduced the
consideration paid by $0.1 million in
working capital adjustments per the purchase agreement. The number of shares to be issued was
determined by the average closing price of our Common Stock as reported on the NYSE Amex for the
ten trading days ending on February 12, 2010. A total of 1,371,428 shares of our Common Stock were
issued in the
60
Table of Contents
transaction which were valued at $2.0 million due to trading restrictions. A portion of the
proceeds will remain in escrow for a period of time to satisfy indemnification claims.
In October 2009, we entered into a definitive agreement to acquire OPKO Chile (previously
known as Pharma Genexx, S.A.), a privately-owned Chilean company engaged in the representation,
importation, commercialization and distribution of pharmaceutical products, over-the-counter
products and medical devices for government, private and institutional markets in Chile. Pursuant
to a stock purchase agreement with OPKO Chile and its shareholders, Farmacias Ahumada S.A., FASA
Chile S.A., and Laboratorios Volta S.A., we acquired all of the outstanding stock of OPKO Chile in
exchange for $16 million in cash. A portion of the proceeds will remain in escrow for a period of
time to satisfy indemnification claims. The transaction closed on October 7, 2009.
The following table summarizes the estimated fair value of the net assets acquired and
liabilities assumed in the acquisition of OPKO Chile at the date of acquisition:
| (in thousands) | ||||
Current assets (including cash of $368) |
$ | 12,208 | ||
Intangible assets |
7,826 | |||
Goodwill |
4,983 | |||
Other assets |
20 | |||
Accounts payable and accrued expenses |
(9,037 | ) | ||
Total purchase price |
$ | 16,000 | ||
Investments
In November 2010, we made an investment in Fabrus, LLC, a privately held early stage
biotechnology company with next generation therapeutic antibody drug discovery and development
capabilities. Fabrus is using its proprietary antibody screening and engineering approach to
discover promising lead compounds against several important oncology targets. In exchange for the
investment, we acquired approximately 13% of Fabrus outstanding membership interests on a fully
diluted basis. Our investment was part of a $2.1 million financing for Fabrus and included other
related parties. Refer to Note 11.
Effective September 21, 2009, we entered into an agreement pursuant to which we invested $2.5
million in cash in Cocrystal Discovery, Inc., a privately held biopharmaceutical company
(Cocrystal) in exchange for 1,701,723 shares of Cocrystals Convertible Series A Preferred Stock
or approximately 16%. Cocrystal is focused on the discovery and development of novel antiviral
drugs using a combination of protein structure-based approaches. Refer to Note 11.
On June 10, 2009, we entered into a stock purchase agreement with Sorrento Therapeutics, Inc.
(Sorrento), a publicly held company with a technology for generating fully human monoclonal
antibodies, pursuant to which we invested $2.3 million in Sorrento. OPKO owns approximately
53,113,732 shares of Sorrento Common Stock, or approximately 21% of Sorrentos total outstanding
common stock at December 31, 2010. The closing stock price for Sorrentos common stock, a thinly
traded stock, as quoted on the over-the-counter markets was $0.60 per share on December 31, 2010.
Refer to Note 11.
The
following table reflects our maximum exposure to each of our investments:
| Investee name | (in thousands) | Accounting method | ||||
Sorrento |
$ | 2,300 | Equity method | |||
Cocrystal |
2,500 | VIE, equity method | ||||
Fabrus |
650 | VIE, equity method | ||||
TESARO |
731 | VIE, cost method | ||||
Less accumulated losses in investees |
(1,067 | ) | ||||
Total |
$ | 5,114 | ||||
Other acquisition
On May 6, 2008, we completed the acquisition of Vidus Ocular, Inc. (Vidus), a privately-held
company that is developing Aquashunt, a shunt to be used in the treatment of glaucoma. Pursuant
to a Securities Purchase Agreement with Vidus, each of its stockholders, and the holders of
convertible promissory notes issued by Vidus, we acquired all of the outstanding stock and
convertible debt of Vidus in exchange for (i) the issuance and delivery at closing of 658,080
shares of our common stock (the Closing Shares); (ii) the issuance of 488,420 shares of our
common stock to be held in escrow pending the occurrence of certain development milestones (the
Milestone Shares); and (iii) the issuance of options to acquire 200,000 shares of our common
stock. Additionally, in the event that the stock price for our common stock at the time of receipt
of approval or clearance by the U.S. Food & Drug Administration of a pre-market notification 510(k)
relating to the Aquashunt is not at or above a specified price, we will be obligated to issue an
additional 413,850 shares of our common stock. A portion of the Closing Shares and the Milestone
Shares remained in escrow for a period of one year to satisfy indemnification claims.
We accounted for the Vidus acquisition as an asset acquisition. We valued the common stock
issued to Vidus stockholders at the average closing price on the date of the acquisition and the
two days prior to the transaction, or
61
Table of Contents
$1.65 per share. In addition, we valued the options to acquire our common stock that were
issued to the founders of Vidus using the Black-Scholes-Merton pricing model and recorded the value
of those options as part of the purchase price of Vidus, or $1.17 per common stock option. All
other contingent consideration will be valued and added to the purchase price if the milestones
occur.
The table below reflects the estimated fair value of the net assets acquired at the date of
acquisition:
| (in thousands) | ||||
Current assets (cash of $48) |
$ | 48 | ||
In-process research and development |
1,398 | |||
Accounts payable and accrued expenses |
(127 | ) | ||
Total purchase price |
$ | 1,319 | ||
The portion of the purchase price allocated to in-process research and development of $1.4
million was immediately expensed.
Variable interest entities
We have determined that we hold variable interests in three entities (VIE), TESARO, Fabrus
and CoCrystal. We made this determination as a result of our assessment that they do not have
sufficient resources to carry out their principal activities without additional subordinated
financial support.
In order to determine the primary beneficiary of Cocrystal and Fabrus, we evaluated our
investment as well as our investment combined with a related party group to identify who had the
most power to control each entity and who received the largest benefits (absorbed the most losses)
from each entity. The related party group when considering our investment in Cocrystal includes
OPKO and the Frost Group. As of December 31, 2010 we own approximately 16% of Cocrystal and
members of the Frost Group own approximately 42% of Cocrystals voting stock on an as converted
basis, including 39% held by the Gamma Trust. Dr. Frost, Mr. Rubin, and Dr. Hsiao currently serve
on the Board of Directors of Cocrystal and represent 50% of its board. The Gamma Trust influenced
the design of Cocrystal and can significantly influence the success of Cocrystal through its board
representation and voting power. As such, we have determined that the Gamma Trust is the primary
beneficiary within the related party group. The related party group when considering our
investment in Fabrus includes OPKO and the Gamma Trust, Hsu Gamma Investment, L.P., of which Jane
Hsiao is the general partner, and the Richard Lerner Family Trust. Dr.s Frost, Hsiao and Lerner
are all members of our Board of Directors. As of December 31, 2010 we own approximately 13% of
Fabrus and Dr.s Frost, Hsiao and Lerner own 24% of Fabrus voting stock on an as converted basis,
including 16% held by the Gamma Trust. Dr.s Frost and Hsiao currently serve on the Board of
Managers of Fabrus and represent 40% of its board. The Gamma Trust can significantly influence the
success of Fabrus through its board representation and voting power. As such, we have determined
that the Gamma Trust is the primary beneficiary within the related
party group. Because we have
the ability to exercise significant influence over Cocrystals
and Fabrus operations through our related party affiliates, we account for our
investments in Cocrystal and Fabrus, under the equity method.
In order to determine the primary beneficiary of TESARO, we evaluated the power and benefits
held by its equity holders. On an as converted basis, we hold an equity interest of approximately
9% of TESARO as of December 31, 2010. In addition, we do not hold any seats on the Board of
Directors and we do not have any management positions. The largest equity holder owns
approximately 49% of TESARO, on an as converted basis and is represented by two members of TESAROs
board of directors. As a result of that equity holder having the power to influence TESARO and
being entitled to the largest share of the benefits of TESARO, we determined such holder is the
primary beneficiary of TESARO. Because we do not have the ability to exercise
significant influence over TESAROs operations, we have
accounted for TESARO under the cost method of accounting.
62
Table of Contents
We have not provided financial or other support to the variable interest entities other than
those associated with our original investments in Cocrystal and Fabrus or those associated with our
TESARO License and we are not obligated to provide ongoing financial
support to them.
Pro forma disclosures for acquisitions
The following table includes the pro forma results for the years ended December 31, 2009 and
2008 of the combined companies as though the acquisition of OPKO Chile had been completed as of the
beginning of each period, respectively.
| For the year ended December 31, | ||||||||
| (in thousands, except per share amounts) | 2009 | 2008 | ||||||
Revenue |
$ | 25,615 | $ | 20,365 | ||||
Net loss |
$ | (28,443 | ) | $ | (39,713 | ) | ||
Basic and diluted loss per share |
$ | (0.12 | ) | $ | (0.21 | ) | ||
This unaudited pro forma financial information is presented for informational purposes only.
The unaudited pro forma financial information may not necessarily reflect our future results of
operations or what the results of operations would have been had we owned and operated each company
as of the beginning of the periods presented.
Note 3 Summary of Significant Accounting Policies
Basis of Presentation. The accompanying consolidated financial statements have been prepared
in accordance with accounting principles generally accepted in the United States and with the
instructions to Form 10-K and of Regulation S-X. The consolidated financial statements include the
accounts of the Company and its wholly-owned subsidiaries. All significant intercompany
transactions and balances have been eliminated in consolidation.
Use of Estimates. The preparation of financial statements in conformity with accounting
principles generally accepted in the United States of America requires management to make estimates
and assumptions that affect the reported amounts of assets and liabilities and disclosure of
contingent assets and liabilities at the date of the financial statements and the reported amounts
of revenues and expenses during the reporting period. Actual results could differ from those
estimates.
Cash and Cash Equivalents. We consider all non-restrictive, highly liquid short-term
investments purchased with a maturity of three months or less on the date of purchase to be cash
equivalents.
Inventories. Inventories are valued at the lower of cost or market (net realizable value).
Cost is determined by the first-in, first-out method.
Property and Equipment. Property and equipment are recorded at cost. Depreciation is
provided using the straight-line method over the estimated useful lives of the assets, generally
five to ten years and includes amortization expense for assets capitalized under capital leases.
The estimated useful lives by asset class are as follows: software 3 years, machinery and
equipment 5-8 years, furniture and fixtures 5-10 years and leasehold improvements the
lesser of their useful life or the lease term. Expenditures for repairs and maintenance are
charged to expense as incurred, while betterments reduce accumulated depreciation. Depreciation
expense was $0.3 million, $0.2 million, and $0.1 million for the years ended December 31, 2010,
2009, and 2008, respectively.
Goodwill and Intangible Assets. Goodwill represents the difference between the purchase price
and the estimated fair value of the net assets acquired when accounted for by the purchase method
of accounting and arose from our acquisitions of OPKO Chile, Exakta-OPKO, and OTI. Refer to Note
2. We do not amortize goodwill, however, we perform an annual impairment test of goodwill during
the fourth quarter. During the fourth quarter of
63
Table of Contents
2009, we performed an impairment test and determined the $1.1 million goodwill related to our
instrumentation business was impaired and written down to $0. As a
result of competition in the U.S. market, the broad global economic
conditions, and pricing pressures globally, we determined that
goodwill was impaired for the instrumentation reporting unit. The
impairment loss was determined by calculating the fair value of the
instrumentation reporting unit based on a discounted net
present-value calculation. We did not record any impairments
during 2010. We evaluate our goodwill for impairment annually and whenever events and changes in
circumstances suggest that the carrying amount may not be recoverable.
We amortize intangible assets with definite lives on a straight-line basis over their
estimated useful lives, ranging from 3 to 10 years, and review for impairment at least annually, or
sooner when events or changes in circumstances indicate that the carrying amount of such assets may
not be recoverable. We use the straight-line method of amortization as there is no reliably
determinable pattern in which the economic benefits of our intangible assets are consumed or
otherwise used up. Amortization expense was $3.6 million, $2.1 million, and $1.7 million for the
years ended December 31, 2010, 2009, and 2008, respectively. In addition, the 2010 and 2009 years
include amortization related to the acquisition of OPKO Chile and the 2010 year end includes
amortization related to our acquisition of Exakta-OPKO. Amortization expense for our intangible
assets as of December 31, 2010 for the years ending December 31, 2011, 2012, 2013, 2014, and 2015
is expected to be $2.2 million, $1.8 million, $0.8 million, $0.8 million, and $0.8 million,
respectively.
Impairment of Long-Lived Assets. Long-lived assets, such as property and equipment, are
reviewed for impairment whenever events or changes in circumstances indicate that the carrying
amount of an asset may not be recoverable. Recoverability of assets to be held and used is
measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash
flows expected to be generated by the asset. If the carrying amount of an asset exceeds its
estimated future cash flows, then an impairment charge is recognized by the amount by which the
carrying amount of the asset exceeds the fair value of the asset.
Fair Value Measurements. The carrying amounts of our cash and cash equivalents, accounts
receivable, accounts payable, and accrued expenses approximate their fair value due to the
short-term maturities of these instruments. Investments are considered available-for-sale as of
December 31, 2010 and 2009, and are carried at fair value.
Short-term investments include bank deposits, corporate notes, U.S. treasury securities and
U.S. government agency securities with original maturities of greater than 90 days and remaining
maturities of less than one year. Long-term investments include corporate notes, U.S. treasury
securities and U.S. government agency securities with maturities greater than one year.
In evaluating the fair value information, considerable judgment is required to interpret the
market data used to develop the estimates. The use of different market assumptions and/or
different valuation techniques may have a material effect on the estimated fair value amounts.
Accordingly, the estimates of fair value presented herein may not be indicative of the amounts that
could be realized in a current market exchange. Refer to Note 17.
Derivative financial instruments. We record derivative financial instruments (primarily
forward purchase contracts) on our balance sheet at their fair value and the changes in the fair
value are recognized in income when they occur, the only exception being derivatives that qualify
as hedges. To qualify the derivative instrument as a hedge, we are required to meet strict hedge
effectiveness and contemporaneous documentation requirements at the initiation of the hedge and
assess the hedge effectiveness on an ongoing basis over the life of the hedge. At December 31,
2010, our forward contracts did not meet the documentation requirements to be designated effective
hedges. Accordingly, we recognize all changes in fair values of our forward contracts in income.
Research and Development. Research and development costs are charged to expense as incurred.
We record expense for in-process research and development projects acquired as asset acquisitions
which have not reached technological feasibility and which have no alternative future use. For
in-process research and development projects acquired in business combinations, the in-process
research and development project is capitalized and evaluated for impairment until the development
process has been completed. Once the development process has been completed the asset will be
amortized over its remaining useful life.
Income Taxes. Income taxes are accounted for under the asset-and-liability method. Deferred
tax assets and liabilities are recognized for the future tax consequences attributable to
differences between the financial statement carrying amounts of existing assets and liabilities and
the respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and
liabilities are measured using enacted tax rates expected to apply to taxable income in the years
in which those temporary differences are expected to be recovered or settled. The effect on
deferred tax assets and liabilities of a change in tax rates is recognized in operations in the
period that includes the
64
Table of Contents
enactment date. We periodically evaluate the realizability of our net deferred tax assets.
Our tax accruals are analyzed periodically and adjustments are made as events occur to warrant such
adjustment.
Loss Per Common Share. Basic and diluted earnings or loss per common share is based on the
net loss increased by dividends on preferred stock divided by the weighted average number of common
shares outstanding during the period. In the periods in which their effect would be anti-dilutive,
no effect has been given to outstanding options, warrants or convertible preferred stock in the
diluted computation. The diluted loss per share does not include the weighted average impact of
the outstanding options, warrants, and other contingent consideration of 21,213,035, 17,743,032,
and 24,022,713 shares for the years ended December 31, 2010, 2009, and 2008 respectively, because
their inclusion would have been anti-dilutive. As of December 31, 2010, the holders of our Series
A Preferred Stock and Series D Preferred Stock could convert their shares into approximately
987,182 and 13,311,823 shares of our Common Stock, respectively, including accrued dividends.
Revenue Recognition. Generally, we recognize revenue from product sales when goods are
shipped and title and risk of loss transfer to our customers. Certain of our products are sold
directly to end-users and require that we deliver, install and train the staff at the end-users
facility. As a result, we do not recognize revenue until the product is delivered, installed and
training has occurred.
Allowance for Doubtful Accounts. We analyze accounts receivable and historical bad debt
levels, customer credit worthiness and current economic trends when evaluating the adequacy of the
allowance for doubtful accounts using the specific identification method. Our reported net loss is
directly affected by managements estimate of the collectability of accounts receivable. Estimated
allowances for sales returns are based upon our history of product returns. The amount of
allowance for doubtful accounts at December 31, 2010 and 2009 was $1.2 million and $0.4 million,
respectively.
Product Warranties. Product warranties are accrued at the time we record revenue for a
product. The costs of warranties are recorded as a component of cost of sales. We estimate
warranty costs based on our estimated historical experience and adjust for any known product
reliability issues.
Equity-Based Compensation. We measure the cost of employee services received in exchange for
an award of equity instruments based on the grant-date fair value of the award. That cost is
recognized in the statement of operations over the period during which an employee is required to
provide service in exchange for the award. We record excess tax benefits, realized from the
exercise of stock options as a financing cash inflow rather than as a reduction of taxes paid in
cash flow from operations. Refer to Note 8. Equity-based compensation arrangements to
non-employees are recorded at their fair value on the measurement date. The measurement of
equity-based compensation is subject to periodic adjustment as the underlying equity instruments
vest.
Comprehensive income or loss. Our comprehensive loss for the year ended December 31, 2010
includes net loss for the year and the cumulative translation adjustment, net, for the translation
of our OPKO Chile and Exakta-OPKO results. Comprehensive loss for the year ended December 31, 2009
is our net loss for the year and the cumulative translation adjustment, net, for the translation of
our OPKO Chile.
Segment reporting. Our chief operating decision-maker (CODM) is comprised of our executive
management with the oversight of our board of directors. Our CODM review our operating results and
operating plans and make resource allocation decisions on a company-wide or aggregate basis.
Accordingly, we have aggregated our three operating segments, instrumentation, pharmaceutical
operating business and our pharmaceutical and device research and development activities into two
reporting segments, instrumentation and pharmaceutical.
Recent accounting pronouncements: In December 2010, the FASB issued an amendment to the
disclosure of supplementary pro forma information for business combinations. The amendment
specifies that if a public entity presents comparative financial statements, the entity should
disclose revenue and earnings of the combined entity as though the business combination(s) that
occurred during the current year had occurred as of the beginning of the comparable prior annual
reporting period only. The amendment also expands the supplemental pro forma disclosures under
current accounting guidance to include a description of the nature and amount of material,
nonrecurring pro forma adjustments directly attributable to the business combination included in
the reported pro forma revenue and earnings. The amendment is effective prospectively for business
combinations for which the acquisition date is on or after the beginning of the first annual
reporting period beginning on or after December 15, 2010. Early adoption is permitted. The
adoption of this amendment is not expected to have a material impact on our financial statement
disclosures.
65
Table of Contents
In December 2010, the FASB issued an amendment to the accounting for goodwill impairment
tests. The amendment modifies Step 1 of the impairment test for reporting units with zero or
negative carrying amounts. For those reporting units, an entity is required to perform Step 2 of
the goodwill impairment test if it is more likely than not that a goodwill impairment exists. In
determining whether it is more likely than not that a goodwill impairment exists, an entity should
consider whether there are any adverse qualitative factors indicating that an impairment may exist.
The qualitative factors are consistent with the existing guidance. The amendment is effective for
fiscal years, and interim periods within those years, beginning after December 15, 2010. Early
adoption is not permitted. The adoption of this amendment is not expected to have a material
impact on our results of operations or financial condition.
In December 2010, the FASB issued an amendment to the accounting for annual excise taxes paid
to the federal government by pharmaceutical manufacturers under health care reform. The liability
for the fee should be estimated and recorded in full upon the first qualifying branded prescription
drug sale with a corresponding deferred cost that is amortized to expense using a straight-line
method of allocation unless another method better allocates the fee over the calendar year that it
is payable. The amendment is effective for calendar years beginning after December 31, 2010, when
the fee initially becomes effective. As we currently do not
manufacture pharmaceutical products, we do not expect the
adoption of this amendment to have material impact on our results of operations or financial condition.
In March 2010, the FASB reached a consensus to issue an amendment to the accounting for
revenue arrangements under which a vendor satisfies its performance obligations to a customer over
a period of time, when the deliverable or unit of accounting is not within the scope of other
authoritative literature, and when the arrangement consideration is contingent upon the achievement
of a milestone. The amendment defines a milestone and clarifies whether an entity may recognize
consideration earned from the achievement of a milestone in the period in which the milestone is
achieved. This amendment is effective for fiscal years beginning on or after June 15, 2010, with
early adoption permitted. The amendment may be applied retrospectively to all arrangements or
prospectively for milestones achieved after the effective date. We have not adopted this guidance
early and adoption of this amendment is not expected to have a material impact on our results of
operation or financial condition.
In January 2010, the FASB issued an amendment to the accounting for fair value measurements
and disclosures. This amendment details additional disclosures on fair value measurements,
requires a gross presentation of activities within a Level 3 rollforward, and adds a new
requirement to disclose transfers in and out of Level 1 and Level 2 measurements. The new
disclosures are required of all entities that are required to provide disclosures about recurring
and nonrecurring fair value measurements. This amendment is effective in the first interim or
reporting period beginning after December 15, 2009, with an exception for the gross presentation of
Level 3 rollforward information, which is required for annual reporting periods beginning after
December 15, 2010, and for interim reporting periods within those years. The adoption of this
amendment is not expected to have a material impact on our financial statement disclosures.
In October 2009, the FASB issued an amendment to the accounting for multiple-deliverable
revenue arrangements. This amendment provides guidance on whether multiple deliverables exist, how
the arrangements should be separated, and how the consideration paid should be allocated. As a
result of this amendment, entities may be able to separate multiple-deliverable arrangements in
more circumstances than under existing accounting guidance. This guidance amends the requirement
to establish the fair value of undelivered products and services based on objective evidence and
instead provides for separate revenue recognition based upon managements best estimate of the
selling price for an undelivered item when there is no other means to determine the fair value of
that undelivered item. The existing guidance previously required that the fair value of the
undelivered item reflect the price of the item either sold in a separate transaction between
unrelated third parties or the price charged for each item when the item is sold separately by the
vendor. If the fair value of all of the elements in the arrangement was not determinable, then
revenue was deferred until all of the items were delivered or fair value was determined. This
amendment will be effective prospectively for revenue arrangements entered into or materially
modified in fiscal years beginning on or after June 15, 2010. Early adoption and retrospective
application is also permitted. We are currently evaluating the potential effect of the adoption of
this amendment on our results of operations or financial condition.
66
Table of Contents
Note 4 Composition of Certain Financial Statement Captions
| December 31, | ||||||||
| (in thousands) | 2010 | 2009 | ||||||
Accounts receivable, net |
||||||||
Accounts receivable |
$ | 14,482 | $ | 9,118 | ||||
Less allowance for doubtful accounts |
(1,165 | ) | (351 | ) | ||||
| $ | 13,317 | $ | 8,767 | |||||
Inventories, net |
||||||||
Raw materials (components) |
$ | 4,868 | $ | 3,764 | ||||
Work in-process |
889 | 1,365 | ||||||
Finished products |
14,632 | 5,632 | ||||||
Less inventory reserve |
(432 | ) | (241 | ) | ||||
| $ | 19,957 | $ | 10,520 | |||||
Prepaid expenses and other current assets |
||||||||
Prepaid supplies and clinical |
$ | 96 | $ | 559 | ||||
Other receivables |
675 | 441 | ||||||
Prepaid insurance |
119 | 162 | ||||||
Taxes recoverable |
1,441 | 414 | ||||||
Other |
451 | 297 | ||||||
| $ | 2,782 | $ | 1,873 | |||||
Property and equipment, net |
||||||||
Machinery and equipment |
$ | 2,406 | $ | 388 | ||||
Building |
288 | | ||||||
Land |
495 | | ||||||
Furniture and fixtures |
179 | 207 | ||||||
Software |
609 | 207 | ||||||
Leasehold improvements |
109 | 237 | ||||||
Less accumulated depreciation |
(1,357 | ) | (446 | ) | ||||
| $ | 2,729 | $ | 593 | |||||
Intangible assets, net |
||||||||
Customer relationships |
$ | 7,719 | $ | 7,259 | ||||
Product registrations |
4,227 | 3,829 | ||||||
Technology |
4,597 | 4,597 | ||||||
Tradename |
666 | 578 | ||||||
Covenants not to compete |
349 | 317 | ||||||
Other |
7 | 7 | ||||||
Less accumulated amortization |
(7,601 | ) | (3,865 | ) | ||||
| $ | 9,964 | $ | 12,722 | |||||
Accrued expenses |
||||||||
Income taxes payable |
$ | 422 | $ | 492 | ||||
Accrued royalties |
316 | 315 | ||||||
Accrued distributor commissions |
398 | 372 | ||||||
Product warranties medical device products |
512 | 257 | ||||||
Clinical trials |
801 | 163 | ||||||
Customer deposits |
321 | 307 | ||||||
Professional fees |
288 | 223 | ||||||
Employee benefits |
304 | 340 | ||||||
Suppliers |
1,240 | | ||||||
Other |
1,137 | 1,449 | ||||||
| $ | 5,739 | $ | 3,918 | |||||
67
Table of Contents
The
following table summarizes the fair values assigned to our major intangible asset classes
upon acquisition:
| Fair value | Weighted average | |||||||
| (in thousands) | assigned | amortization period | ||||||
Customer relationships |
$ | 7,797 | 3 years | |||||
Technology |
4,597 | 10 years | ||||||
Product registrations |
3,829 | 10 years | ||||||
Covenants not to compete |
366 | 3 years | ||||||
Tradename |
666 | 3 years | ||||||
Other |
7 | Indefinite | ||||||
Total amortizing intangible assets |
17,262 | |||||||
Goodwill |
5,408 | Indefinite | ||||||
Total intangible assets acquired |
$ | 22,670 | ||||||
All of the intangible assets and goodwill acquired relate to our acquisitions of OPKO Chile,
Exakta-OPKO, and OTI. The weighted average period prior to the next renewal or extension for our
product registrations is 2.7 years. We do not anticipate capitalizing the cost of product
registration renewals, rather we expect to expense these costs, as incurred. Our goodwill is not tax
deductible for income tax purposes in Chile.
The changes to goodwill for the year ended December 31, 2010 is primarily due to a $0.4
million increase resulting from foreign exchange translation of the assets and liabilities of OPKO
Chile. The purchase price allocation of the assets acquired in the Exakta-OPKO acquisition are
subject to change while contingencies that existed on the acquisition date are resolved.
The following table reflects the changes in the allowance for doubtful accounts, provision for
inventory reserve and tax valuation allowance accounts:
| Beginning | Charged to | Charged to | Ending | |||||||||||||||||
| (in thousands) | balance | expense | Written-off | other | balance | |||||||||||||||
2010 |
||||||||||||||||||||
Allowance for doubtful accounts |
$ | (351 | ) | (446 | ) | | (368 | ) | $ | (1,165 | ) | |||||||||
Inventory reserve |
$ | (241 | ) | (64 | ) | 170 | (297 | ) | $ | (432 | ) | |||||||||
Tax valuation allowance |
$ | (51,697 | ) | (2,555 | ) | | | $ | (54,252 | ) | ||||||||||
2009 |
||||||||||||||||||||
Allowance for doubtful accounts |
$ | (407 | ) | (73 | ) | 129 | | $ | (351 | ) | ||||||||||
Inventory reserve |
$ | (255 | ) | (279 | ) | 293 | | $ | (241 | ) | ||||||||||
Tax valuation allowance |
$ | (35,197 | ) | (16,699 | ) | | 199 | $ | (51,697 | ) | ||||||||||
Note 5 Debt
We have a $12.0 million line of credit with the Frost Group, a related party. On June 2, 2010
we repaid all amounts outstanding on the line of credit including $12 million in principal and $4.1
million in interest. The line of credit was renewed on February 22, 2011 with a new maturity date
of March 31, 2012. We have the ability to draw funds under the line of credit until its expiration
in March 2012. We are obligated to pay interest upon maturity, capitalized quarterly, on
outstanding borrowings under the line of credit at an 11% annual rate. The line of credit is
collateralized by all of our U.S. personal property except our intellectual property.
We have entered into lines of credit agreements with seven financial institutions in Chile in
addition to our line of credit with the Frost Group. Those lines of credit are used primarily as a
source of working capital for inventory purchases. The following table summarizes the lines of
credit:
68
Table of Contents
| (in thousands) | Interest rate on | Maximum | Amount outstanding at December 31, | |||||||||||||
| Lender | borrowings | borrowings | 2010 | 2009 | ||||||||||||
The Frost Group LLC |
11% | $ | 12,000 | $ | | $ | 12,000 | |||||||||
Itau Bank |
Libor +2.8% | 3,000 | 1,849 | 270 | ||||||||||||
Bank of Chile |
Libor +2.8% | 3,000 | 3,100 | 988 | ||||||||||||
BICE Bank |
Libor +2.8% | 3,300 | 2,813 | 1,459 | ||||||||||||
Santander Bank |
Libor +2.8% | 2,500 | 1,826 | 324 | ||||||||||||
Corp Banca |
Libor +2.8% | 1,050 | 426 | 62 | ||||||||||||
BBVA Bank |
Libor +2.8% | 3,500 | 3,123 | 1,218 | ||||||||||||
Scotiabank |
Libor +2.8% | 2,500 | 1,553 | | ||||||||||||
Total |
$ | 30,850 | $ | 14,690 | $ | 16,321 | ||||||||||
On March 4, 2009, the Gamma Trust, a related party, advanced $3.0 million to us pursuant to a
Promissory Note we issued to the Gamma Trust (the Note). The entire amount of this advance and
all accrued interest thereon was due and payable on the earlier of May 4, 2009, or such earlier
date following the closing of the stock purchase transaction with the Gamma Trust discussed in Note
6. The Note bore interest at a rate equal to 11% per annum and could be prepaid in whole or in
part without penalty or premium. We repaid the Note and $48 thousand of interest on April 27,
2009.
Note 6 Equity Offerings
Effective September 18, 2009, we entered into a securities purchase agreement (the Preferred
Purchase Agreement) with the private investors (the Preferred Investors), pursuant to which the
Preferred Investors agreed to purchase an aggregate of 1,209,677 shares (the Preferred Shares) of
our newly-designated 8.0% Series D Cumulative Convertible Preferred Stock, par value $0.01 per
share (Series D Preferred Stock) (Refer to Note 7) at a purchase price of $24.80 per share,
together with warrants (the Warrants) to purchase up to an aggregate of 3,024,196 shares of our
common stock, par value $.01 at an exercise price of $2.48 per share (the Preferred Investment).
Initially, the Series D Preferred Stock was convertible into ten shares of our Common Stock, and
the Preferred Shares purchase price was based on the average closing price of our Common Stock as
reported on the NYSE Amex for the five days preceding the execution of the Preferred Purchase
Agreement. In connection with the Preferred Investment, we issued the Preferred Shares and
Warrants and received an aggregate of $30.0 million in cash on September 28, 2009.
We allocated the $30.0 million of proceeds from the Preferred Investment between the Series D
Preferred Stock and the Warrants based on their relative fair values as follows:
| (in thousands) | ||||
Series D Preferred Stock |
$ | 26,128 | ||
Warrants Settlements in kind or expired |
3,872 | |||
Total |
$ | 30,000 | ||
We allocated the $30 million in proceeds received from the issuance of the Preferred Stock and
warrants to those instruments based on their relative fair values, which resulted in a $3.9 million
beneficial conversion feature. We recorded the $3.9 million beneficial conversion feature as a
further discount to the Series D Preferred Stock and an increase to additional paid-in capital.
The Series D Preferred Stock was immediately convertible into shares of our common stock. As
a result, the discount was immediately recognized as a deemed dividend and included in preferred
stock dividends in the accompanying consolidated statement of operations. The Series D Preferred
Stock contains redemption features that are not solely within our control. As a result, the Series
D Preferred Stock is classified outside of permanent equity. The Series D Preferred Stock is
recorded at this time at initial fair value and not at its Liquidation Amount as it is not probable
that it will be redeemed.
We agreed to issue the Preferred Shares and the Warrants in reliance upon the exemption from
registration under Section 4(2) of the Securities Act of 1933, as amended (the Act). The
Preferred Shares issued in the Preferred Investment, including the shares of the Companys Common
Stock into which the Preferred Shares and Warrants may be converted, are restricted securities as
that term is defined by Rule 144 under the Act, subject to a three year contractual lockup, and no
registration rights have been granted.
69
Table of Contents
On May 26, 2009, May 29, 2009, and June 1, 2009, we entered into stock purchase agreements
with a total of seven accredited investors (Investors) pursuant to which the Investors agreed to
make a $31.0 million investment in the Company in exchange for 31,000,000 shares of our Common
Stock at $1.00 per share, representing a range of discounts of approximately 16-21% to the average
closing price of our Common Stock on the NYSE Amex for the five trading days immediately preceding
the closing date of the agreements. The shares issued were restricted securities and were exempt
from registration requirements under Section 4(2) of the Act because the transaction did not
involve a public offering.
On February 23, 2009, we entered into a Stock Purchase Agreement with the Gamma Trust,
pursuant to which the Gamma Trust agreed to make a $20.0 million cash investment in the Company in
exchange for 20,000,000 shares (the Shares) of our Common Stock, at $1.00 per share, representing
an approximately 20% discount to the average closing price of our Common Stock on the NYSE Amex for
the five trading days immediately preceding the effective date of Audit Committee and stockholder
approval of the transaction. We issued the Shares and received the proceeds on April 27, 2009.
The Shares issued were restricted securities, subject to a two-year lockup and no registration
rights were granted.
On September 10, 2008, we issued 13,513,514 shares of our Common Stock to a group of
investors, including members of the Frost Group, in exchange for $15.0 million. The shares were
issued at $1.11 per share, representing an approximately 40% discount to the five-day average
closing price of our Common Stock on the NYSE Amex. The shares issued were restricted securities,
subject to a two year lockup, and no registration rights have been granted. The issuance of the
shares was exempt from the registration requirements under Section 4(2) of the Act because the
transaction did not involve a public offering.
Note 7 Shareholders Equity
Our authorized capital stock consists of 500,000,000 shares of common stock, par value $.01
per share, and 10,000,000 shares of preferred stock, par value $.01 per share.
Common Stock
Subject to the rights of the holders of any shares of preferred stock currently outstanding or
which may be issued in the future, the holders of the common stock are entitled to receive
dividends from our funds legally available when, as and if declared by our board of directors, and
are entitled to share ratably in all of our assets available for distribution to holders of common
stock upon the liquidation, dissolution or winding-up of our affairs subject to the liquidation
preference, if any, of any then outstanding shares of preferred stock. Holders of our common stock
do not have any preemptive, subscription, redemption or conversion rights. Holders of our common
stock are entitled to one vote per share on all matters which they are entitled to vote upon at
meetings of stockholders or upon actions taken by written consent pursuant to Delaware corporate
law. The holders of our common stock do not have cumulative voting rights, which means that the
holders of a plurality of the outstanding shares can elect all of our directors. All of the shares
of our common stock currently issued and outstanding are fully-paid and nonassessable. No
dividends have been paid to holders of our common stock since our incorporation, and no cash
dividends are anticipated to be declared or paid in the reasonably foreseeable future.
In addition to our equity-based compensation plans, we have issued warrants to purchase our
common stock. Refer to Note 8 for additional information on our share-based compensation plans.
The table below provides additional information for warrants outstanding as of December 31, 2010.
| Weighted | ||||||||||||
| Number of | average | |||||||||||
| Warrants | warrants | exercise price | Expiration date | |||||||||
Outstanding at December 31, 2009 |
29,194,162 | |||||||||||
Issued |
| |||||||||||
Exercised |
| |||||||||||
Expired |
| |||||||||||
Outstanding and Exercisable at December 31, 2010 |
Various from September 2014 | |||||||||||
| 29,194,162 | $ | 0.89 | through March, 2017 | |||||||||
70
Table of Contents
Preferred Stock
Under our certificate of incorporation, our board of directors has the authority, without
further action by stockholders, to designate up to 10 million shares of preferred stock in one or
more series and to fix or alter, from time to time, the designations, powers and rights of each
series of preferred stock and the qualifications, limitations or restrictions of any series of
preferred stock, including dividend rights, dividend rate, conversion rights, voting rights, rights
and terms of redemption (including sinking fund provisions), redemption price or prices, and the
liquidation preference of any wholly issued series of preferred stock, any or all of which may be
greater than the rights of the common stock, and to establish the number of shares constituting any
such series.
Series A Preferred Stock
Of the authorized preferred stock, 4,000,000 shares have been designated Series A preferred
stock. Dividends are payable on the Series A preferred stock in the amount of $0.25 per share,
payable annually in arrears. At the option of our board of directors, dividends will be paid
either (i) wholly or partially in cash or (ii) in newly issued shares of Series A preferred stock
valued at $2.50 per share to the extent cash dividend is not paid.
Holders of Series A preferred stock have the right to convert their shares, at their option
exercisable at any time, into shares of our common stock on a one-for-one basis subject to
anti-dilution adjustments. These anti-dilution adjustments are triggered in the event of any
subdivision or combination of our outstanding common stock, any payment by us of a stock dividend
to holders of our common stock or other occurrences specified in the certificate of designations
relating to the Series A preferred stock. We may elect to convert the Series A preferred stock
into common stock or a substantially equivalent preferred stock in the case of a merger or
consolidation in which we do not survive, a sale of all or substantially all of our assets or a
substantial reorganization of us.
Each share of Series A preferred stock is entitled to one vote on all matters on which the
common stock has the right to vote. Holders of Series A preferred stock are also entitled to vote
as a separate class on any proposed adverse change in the rights, preferences or privileges of the
Series A preferred stock and any increase in the number of authorized shares of Series A preferred
stock. In the event of any liquidation or winding up of the Company, the holders of the Series A
preferred stock will be entitled to receive $2.50 per share plus any accrued and unpaid dividends
before any distribution to the holders of the common stock and any other class of series of
preferred stock ranking junior to it.
We may redeem the outstanding shares of Series A preferred stock for $2.50 per share (plus
accrued and unpaid dividends), at any time.
Series C Preferred Stock
Of the authorized preferred stock, 500,000 shares were designated Series C preferred stock.
On June 22, 2007, 457,603 shares of Series C preferred stock were issued and outstanding and held
by 30 stockholders. Cumulative dividends were payable on the Series C preferred stock in the
amount of $1.54 per share when declared by the board of directors. On June 22, 2007, all
outstanding shares (457,603 shares) of Series C preferred stock automatically converted into shares
of common stock, on a one-hundred-for-one basis.
8% Series D Cumulative Convertible Preferred Stock
Of the authorized preferred stock, 2,000,000 shares were designated 8% Series D Cumulative
Convertible Preferred Stock (Series D Preferred Stock). Holders of the Series D Preferred Stock
are entitled to receive, when, as and if declared by the Companys Board of Directors, dividends on
each share of Series D Preferred Stock at a rate per annum equal to 8.0% of the sum of (a) $24.80,
plus (b) any and all declared and unpaid and accrued dividends thereon, subject to adjustment for
any stock split, combination, recapitalization or other similar corporate action (the Liquidation
Amount). All dividends shall be cumulative, whether or not earned or declared, accruing on an
annual basis from the issue date of the Series D Preferred Stock. As of December 31, 2010 we had
approximately $2.49 per Series D Preferred Share, or $3.0 million of Series D Preferred Stock
dividends in arrears.
The Holders of Series D Preferred Stock have the right to receive notice of any meeting of
holders of our Common Stock or Series D Preferred Stock and to vote (on an as-converted into Common
Stock basis) upon any matter submitted to a vote of the holders of Common Stock or Series D
Preferred Stock. Except as otherwise expressly set forth in the Companys Amended and Restated
Certificate of Incorporation, as amended from time to time, the holders of Series D Preferred Stock
will vote on each matter submitted to them with the holders of
71
Table of Contents
Common Stock and all other classes and series of our capital stock entitled to vote on such
matter, taken together as a single class.
With respect to dividend distributions (other than required dividends to the holders of our
Series A Preferred Stock) and distributions upon liquidation, winding up or dissolution of the
Company, the Series D Preferred Stock ranks senior to all classes of Common Stock, our Series A
Preferred Stock, our Series C Preferred Stock, and to each other class of our capital stock
existing now or hereafter created that are not specifically designated as ranking senior to or pari
passu with the Series D Preferred Stock.
Upon the occurrence of a Liquidation Event (as defined in the Certificate of Designation),
holders of Series D Preferred Stock are entitled to be paid, subject to applicable law, out of the
assets of the Company available for distribution to its stockholders, an amount in cash (the
Liquidation Payment) for each share of Series D Preferred Stock equal to the greater of (x) the
Liquidation Amount for each such share of Series D Preferred Stock outstanding plus (i) any
declared and unpaid dividends and (ii) accrued dividends or (y) the amount for each share of Series
D Preferred Stock the holders would be entitled to receive pursuant to the Liquidation Event if all
of the shares of Series D Preferred Stock had been converted into Common Stock as of the date
immediately prior to the date fixed for determination of stockholders entitled to receive a
distribution in such Liquidation Event. Such Liquidation Payment will be paid before any cash
distribution will be made or any other assets distributed in respect of any class of securities
junior to the Series D Preferred Stock, including, without limitation, Common Stock and the
Companys Series A Preferred Stock.
The holder of any share of Series D Preferred Stock may at any time and from time to time
convert such share into such number of fully paid and nonassessable shares of Common Stock as is
determined by dividing (A) the Liquidation Amount of the share by (B) the Conversion Price, which
is initially $2.48, subject to adjustment as provided in the Certificate of Designation.
Initially, the Series D Preferred Stock is convertible into 10 shares of the Companys Common
Stock.
We may, at any time, convert the outstanding Series D Preferred Stock into such number of
fully paid and non-assessable shares of Common Stock as is determined by dividing (A) the
Liquidation Amount of the shares by (B) the Conversion Price, but only if the closing bid price of
the Common Stock exceeds $5.00 per share during any thirty (30) consecutive trading days prior to
each conversion. Initially, the Series D Preferred Stock was convertible into 10 shares of the
Companys Common Stock.
To the extent it is lawfully able to do so, we may redeem all of the then outstanding shares
of Series D Preferred Stock by paying in cash an amount per share equal to $24.80 plus all declared
or accrued unpaid dividends on such shares, subject to adjustment for any stock dividends or
distributions, splits, subdivisions, combinations, reclassifications, stock issuances or similar
events with respect to the Common Stock.
Note 8 Equity-Based Compensation
We maintain three equity-based incentive compensation plans, the 2007 Equity Incentive Plan,
the 2000 Stock Option Plan, and the 1996 Stock Option Plan that provide for grants of stock options
and restricted stock to our directors, officers, key employees and certain outside consultants.
Equity awards granted under our 2007 Equity Incentive Plan are exercisable for a period up to seven
years from the date of grant. Equity awards granted under our 2000 Stock Option Plan and the 1996
Stock Option Plan are exercisable for a period of up to 10 years from date of grant. Vesting
periods range from immediate to 5 years.
We classify the cash flows resulting from the tax benefit that arises when the tax deductions
exceed the compensation cost recognized for those equity awards (excess tax benefits) as financing
cash flows. There were no excess tax benefits for the years ended December 31, 2010, 2009, and
2008.
Equity-based compensation arrangements to non-employees are accounted for at their fair value
on the measurement date. The measurement of equity-based compensation is subject to periodic
adjustment over the vesting period of the equity instruments.
Valuation and Expense Information
We recorded equity based compensation expense of $6.9 million, $4.5 million, and $6.7 million
for the years ended December 31, 2010, 2009 and 2008, respectively, all of which were reflected as
operating expenses. Of the $6.9 million of equity based compensation expense recorded in the year
ended December 31, 2010, $5.2 million was recorded as selling, general and administrative expense
and $1.7 million was recorded as research and development
72
Table of Contents
expenses. Of the $4.5 million of equity based compensation expense recorded in the year ended
December 31, 2009, $3.2 million was recorded as selling, general and administrative expense and
$1.3 million was recorded as research and development expenses. For the year ended December 31,
2008, of the $6.7 million of equity based compensation expense recorded, $4.2 million was recorded
as selling, general and administration expense and $2.5 million was recorded as research and
development expense.
We estimate forfeitures of stock options and recognize compensation cost only for those awards
expected to vest. Forfeiture rates are determined for all employees and non-employee directors
based on historical experience and our estimate of future vesting. Estimated forfeiture rates are
adjusted from time to time based on actual forfeiture experience.
As of December 31, 2010, there was $8.2 million of unrecognized compensation cost related to
the stock options granted under our stock plans. That cost is expected to be recognized over a
weighted-average period of approximately 2 years.
Stock Options
We estimate the fair value of each stock option on the date of grant using a Black-Scholes
option-pricing formula, applying the following assumptions, and amortize the fair value to expense
over the options vesting period using the straight-line attribution approach for employees and
non-employee directors, and for awards issued to non-employees we recognize compensation expense on
a graded basis, with most of the compensation expense being recorded during the initial periods of
vesting:
| Year Ended | Year Ended | Year Ended | ||||||||||
| December 31, 2010 | December 31, 2009 | December 31, 2008 | ||||||||||
Expected term (in years) |
0.6 - 7.0 | 0.6 - 7.9 | 1.6 - 8.9 | |||||||||
Risk-free interest rate |
1.3% - 2.7% | 1.4% - 3.0% | 1.5% - 3.7% | |||||||||
Expected volatility |
69% - 74% | 70% - 77% | 70% - 75% | |||||||||
Expected dividend yield |
0% | 0% | 0% | |||||||||
Expected Term: The expected term of the stock options granted to employees and non-employee
directors was calculated using the shortcut method. We believe this method is appropriate as our
equity shares have been publicly traded for a limited period of time and as such we do not have
sufficient historical exercise data to provide a reasonable basis upon which to estimate expected
term. The expected term of stock options issued to non-employee consultants is the remaining
contractual life of the options issued.
Risk-Free Interest Rate: The risk-free interest rate is based on the rates paid on securities
issued by the U.S. Treasury with a term approximating the expected life of the option.
Expected Volatility: The expected volatility was based on a peer group of publicly-traded
stocks historical trading which we believe will be representative of the volatility over the
expected term of the options. We believe the peer groups historical volatility is appropriate as
our equity shares have been publicly traded for a limited period of time.
Expected Dividend Yield: We do not intend to pay dividends on common stock for the foreseeable
future. Accordingly, we used a dividend yield of zero in the assumptions.
We maintain incentive stock plans that provide for the grants of stock options to our
directors, officers, employees and non-employee consultants. As of December 31, 2010, there were
11,106,725 shares of common stock reserved for issuance under our 2007 Incentive Plan. We intend
to issue new shares upon the exercise of options. Stock options granted under these plans have
been granted at an option price equal to the closing market value of the stock on the date of the
grant. Options granted under these plans to employees typically become exercisable over four years
in equal annual installments after the date of grant, and options granted to non-employee directors
become exercisable in full one-year after the grant date, subject to, in each case, continuous
service with the Company during the applicable vesting period. The Company assumed options to
grant common stock as part of the mergers with Acuity Pharmaceuticals,
Inc. and Froptix, Inc., which reflected various vesting schedules, including
monthly vesting to employees and non-employee consultants.
73
Table of Contents
A summary of option activity under our stock plans as of December 31, 2010, and the changes
during the year is presented below:
| Weighted | ||||||||||||||||
| Weighted | average | |||||||||||||||
| average | remaining | Aggregate | ||||||||||||||
| Number of | exercise | contractual | intrinsic value | |||||||||||||
| Options | options | price | term (years) | (in thousands) | ||||||||||||
Outstanding at December 31, 2009 |
12,623,556 | $ | 2.36 | 5.5 | $ | 4,544 | ||||||||||
Granted |
2,736,000 | $ | 2.25 | |||||||||||||
Exercised |
(150,231 | ) | $ | 0.49 | ||||||||||||
Forfeited |
(396,554 | ) | $ | 3.76 | ||||||||||||
Expired |
(104,625 | ) | $ | 3.42 | ||||||||||||
Outstanding at December 31, 2010 |
14,708,146 | $ | 2.31 | 4.8 | $ | 23,464 | ||||||||||
Vested and expected to vest at
December 31, 2010 |
13,852,934 | $ | 2.32 | 4.8 | $ | 22,134 | ||||||||||
Exercisable at December 31, 2010 |
6,775,064 | $ | 2.53 | 4.0 | $ | 10,467 | ||||||||||
The total intrinsic value of stock options exercised for the years ended December 31, 2010,
2009, and 2008 was $0.3 million, $3.8 million, and $9.5 million, respectively.
The weighted average grant date fair value of stock options granted for the years ended
December 31, 2010, 2009 and 2008 was $1.39, $0.99, and $1.13, respectively. The total fair value
of stock options vested during the years ended December 31,
2010, 2009 and 2008 was $3.4 million, $5.1
million, and $4.1 million, respectively. The following table provides the grant date fair value
for each of the following groups of stock option activity during 2010:
| Weighted | ||||||||
| Number of | average grant | |||||||
| Options | options | date fair value | ||||||
Nonvested at December 31, 2009 |
7,516,418 | $ | 1.47 | |||||
Granted |
2,736,000 | $ | 1.39 | |||||
Forfeited |
396,554 | $ | 2.32 | |||||
Nonvested at December 31, 2010 |
7,933,082 | $ | 1.32 | |||||
Restricted Stock
In 2009, we issued 30,000 shares of restricted common stock to one of our independent board
members. The restricted stock was granted under our 2007 Equity Incentive Plan with a term of
seven years and vesting occurring five years after the grant date with certain events which would
accelerate the vesting of the award. The restricted stock was valued using the grant date fair
value which was equivalent to the closing price of our common stock on the grant date. We record
the cost of restricted stock over the vesting period.
74
Table of Contents
Note 9 Income Taxes
We
operate in the following countries in which we are required
to file tax returns: U.S., Canada, Mexico, Taiwan, and Chile.
The (expense) benefit for incomes taxes consists of the following:
| For the year ended December 31, | |||||||||||||
| (in thousands) | 2010 | 2009 | 2008 | ||||||||||
Current |
|||||||||||||
Federal |
$ | | $ | | $ | | |||||||
State |
| | | ||||||||||
Foreign |
(489 | ) | 140 | 83 | |||||||||
| (489 | ) | 140 | 83 | ||||||||||
Deferred |
|||||||||||||
Federal |
| | | ||||||||||
State |
| | | ||||||||||
Foreign |
348 | 154 | | ||||||||||
| 348 | 154 | | |||||||||||
Total, net |
$ | (141 | ) | $ | 294 | $ | 83 | ||||||
Deferred income tax assets and liabilities as of December 31, 2010 and 2009 are comprised of
the following:
| December 31, | December 31, | |||||||
| (in thousands) | 2010 | 2009 | ||||||
Deferred income tax assets: |
||||||||
Federal net operating loss |
$ | 33,489 | $ | 26,690 | ||||
State net operating loss |
3,694 | 4,816 | ||||||
Foreign net operating loss |
1,481 | 1,198 | ||||||
Capitalized research and development expense |
3,677 | 4,378 | ||||||
Research and development tax credit |
2,342 | 6,492 | ||||||
Canadian research and development pool |
1,212 | 1,464 | ||||||
Canadian tax credits |
828 | 1,089 | ||||||
Amortization and depreciation |
298 | 258 | ||||||
Accruals |
19 | 555 | ||||||
Other |
8,898 | 6,663 | ||||||
Deferred income tax assets |
55,938 | 53,603 | ||||||
Deferred income tax liabilities: |
||||||||
Intangible assets |
(2,318 | ) | (3,114 | ) | ||||
Other |
(308 | ) | | |||||
Deferred income tax liabilities |
(2,626 | ) | (3,114 | ) | ||||
Net deferred income tax assets |
53,312 | 50,489 | ||||||
Valuation allowance |
(54,252 | ) | (51,697 | ) | ||||
Net deferred income tax liabilities |
$ | (940 | ) | $ | (1,208 | ) | ||
The change in deferred income tax assets, liabilities and valuation allowances at December 31,
2010 reflect the acquisition of various legal entities, including the tax attributes. The
acquisitions were accounted for under U.S. GAAP as asset acquisitions and business combinations.
As of December 31, 2010, we have federal, state, and foreign net operating loss carryforwards of
approximately $162.7 million, $138.7 million, and $6.0 million, respectively, that expire at
various dates through 2030. We have research and development tax credit carryforwards of
approximately $2.7 million that expire in varying amounts through 2030. We have determined a full
valuation allowance is required against all of our tax assets that we do not expect to be utilized
by the turning of deferred income tax liabilities.
Under Section 382 of the Internal Revenue Code of 1986, as amended, certain significant
changes in ownership may restrict the future utilization of our income tax loss carryforwards and
income tax credit carryforwards in the United States. The annual limitation is equal to the value
of our stock immediately before the ownership change,
75
Table of Contents
multiplied by the long-term tax-exempt rate (i.e., the highest of the adjusted Federal
long-term rates in effect for any month in the three-calendar-month period ending with the calendar
month in which the change date occurs). This limitation may be increased under the IRC§ 338
Approach (IRS approved methodology for determining recognized Built-In Gain). As a result, federal
net operating losses and tax credits may expire before we are able to fully utilize them.
During 2008, we conducted a study to determine the impact of the various ownership changes
that occurred during 2007 and 2008. As a result, we have concluded that the annual utilization of
our net operating loss carryforwards (NOLs) and tax credits is subject to a limitation pursuant
to Internal Revenue Code section 382. Under the tax law, such NOLs and tax credits are subject to
expiration from 15 to 20 years after they were generated. As a result of the annual limitation
that may be imposed on such tax attributes and the statutory expiration period, some of these tax
attributes may expire prior to our being able to use them. As we have established a valuation
allowance against all of our net deferred tax assets, including such NOLs and tax credits, there is
no current impact on these financial statements as a result of the annual limitation. This study
did not conclude as to whether eXegenics pre-merger NOLs were limited under Section 382. As such,
of the $162.7 million of federal net operating loss carryforwards, at least approximately $52.0
million may not be able to be utilized.
Uncertain Income Tax Positions
We
file Federal income tax returns in the U.S., Canada, Chile, Mexico, and Taiwan
jurisdictions, as well as with various U.S. states and the Ontario province in Canada. We are
subject to tax audits in all jurisdictions for which we file tax returns. Tax audits by their very
nature are often complex and can require several years to complete. There are currently no tax
audits that have commenced with respect to income returns in any jurisdiction.
U.S. Federal: Under the tax statute of limitations applicable to the Internal Revenue Code, we
are no longer subject to U.S. federal income tax examinations by the Internal Revenue Service for
years before 2006. However, because we are carrying forward income tax attributes, such as net
operating losses and tax credits from 2006 and earlier tax years, these attributes can still be
audited when utilized on returns filed in the future.
State: Under the statutes of limitation applicable to most state income tax laws, we are no
longer subject to state income tax examinations by tax authorities for years before 2006 in states
in which we have filed income tax returns. Certain states may take the position that we are
subject to income tax in such states even though we have not filed income tax returns in such
states and, depending on the varying state income tax statutes and administrative practices, the
statute of limitations in such states may extend to years before 2006.
Foreign: Under the statutes of limitations applicable to our foreign operations, we are no
longer subject to tax examination for years before 2006 in jurisdictions we have filed income tax
returns.
As a result of our January 1, 2007 implementation of ASC 740, the total amount of gross tax
benefits, excluding the offsetting full valuation allowance, that became unrecognized, was
approximately $0.4 million. There were no accrued interest and penalties resulting from such
unrecognized tax benefits. As of December 31, 2010 and December 31, 2009, the total amount of
gross unrecognized tax benefits was approximately $5.4 million and $6.8 million, respectively, and
accrued interest and penalties on such unrecognized tax benefits was $0 in each period.
The following table rolls forward the 2010 activity in our gross unrecognized income tax
benefits.
| (in thousands) | ||||
Unrecognized tax benefits January 1, 2010 |
$ | 6,818 | ||
Gross increases tax positions in prior period |
| |||
Gross decreases tax positions in prior period |
(1,405 | ) | ||
Unrecognized tax benefits at December 31, 2010 |
$ | 5,413 | ||
There are no net unrecognized tax benefits that, if recognized, would impact the effective tax
rate as of December 31, 2010 as a result of the full valuation allowance.
Other Income Tax Disclosures
The significant elements contributing to the difference between the federal statutory tax rate
and the effective tax rate are as follows:
76
Table of Contents
| For the year ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
Federal statutory rate |
35.0 | % | 35.0 | % | 35.0 | % | ||||||
State income taxes, net of federal
benefit |
3.5 | 3.7 | 3.6 | |||||||||
Foreign income tax |
(1.0 | ) | 0.1 | | ||||||||
Acquired in-process research and
development |
| (2.6 | ) | (1.4 | ) | |||||||
Research and development tax credits |
5.8 | 6.7 | 10.7 | |||||||||
OID |
3.7 | 5.0 | | |||||||||
Impairment of goodwill |
| (1.4 | ) | | ||||||||
Other items
including valuation allowance and permanent items |
(47.0 | ) | (45.5 | ) | (48.0 | ) | ||||||
Other |
(0.8 | ) | 0.0 | 0.3 | ||||||||
Total |
(0.8 | )% | 1.0 | % | 0.2 | % | ||||||
The following table reconciles our losses before income taxes between U.S. and foreign
jurisdictions:
| For the year ended December 31, | ||||||||||||
| (in thousands) | 2010 | 2009 | 2008 | |||||||||
Pre-tax loss |
||||||||||||
U.S. |
$ | (16,256 | ) | $ | (29,214 | ) | $ | (37,153 | ) | |||
Foreign |
(1,815 | ) | (840 | ) | (2,764 | ) | ||||||
Total |
$ | (18,071 | ) | $ | (30,054 | ) | $ | (39,917 | ) | |||
Note 10 Supplemental Cash Flow Information
Supplemental cash flow information is summarized as follows:
| For the year ended December 31, | ||||||||||||
| (in thousands) | 2010 | 2009 | 2008 | |||||||||
Interest paid |
$ | 4,386 | $ | 95 | $ | 101 | ||||||
Income taxes paid, net |
$ | 235 | $ | | $ | | ||||||
Non-cash financing
|
||||||||||||
Issuance of
capital stock to
acquire Exakta-OPKO and Vidus |
$ | 1,999 | $ | | $ | 1,319 | ||||||
Note 11 Related Party Transactions
We have a $12.0 million line of credit with the Frost Group, a related party. On June 2, 2010
we repaid all amounts outstanding on the line of credit including $12 million in principal and $4.1
million in interest. The line of credit, which previously expired on January 11, 2011, was renewed
on February 22, 2011 until March 31, 2012 on substantially the same terms as in effect at the time
of expiration. We have the ability to draw funds under the line of credit until its expiration in
March 2012. We are obligated to pay interest upon maturity, capitalized quarterly, on outstanding
borrowings under the line of credit at an 11% annual rate. The line of credit is collateralized by
all of our U.S. personal property except our intellectual property.
In November 2010, we made an investment in Fabrus, LLC, a privately held early stage
biotechnology company with next generation therapeutic antibody drug discovery and development
capabilities. In exchange for the investment, we acquired approximately 13% of Fabrus outstanding
membership interests on a fully diluted basis. Our investment was
part of a $2.1 million
financing for Fabrus. Other investors participating in the financing include Frost Gamma
Investments Trust, of which Phillip Frost is the sole trustee, and Hsu Gamma Investment, L.P., of
which Jane Hsiao, the Companys Vice Chairman and Chief Technical Officer, serves as the general
partner. In connection with the financing, Drs. Frost and Hsiao joined the Fabrus Board of
Managers. Dr.
77
Table of Contents
Richard Lerner, a director of the Company, owns approximately 5% of Fabrus. Vaughn Smider,
Founder and CEO of Fabrus, is an Assistant Professor at The Scripps Research Institute (TSRI).
Dr. Frost serves as a Trustee for TSRI, and Richard Lerner serves as its President.
On July 20, 2010, we entered into a use agreement for approximately 1,100 square feet of space
in Jupiter, Florida to house our molecular diagnostics operations with TSRI. Dr. Frost is a member
of the Board of Trustees of TSRI and Dr. Richard Lerner, a member of our Board of Directors, is
also the President of TSRI. Pursuant to the terms of the use agreement, which is effective as of
November 1, 2009, gross rent is approximately $40 thousand per year for a two-year term which may
be extended, upon mutual agreement, for one additional year.
On June 1, 2010, the Company entered into a cooperative research and development agreement
with Academia Sinica in Taipei, Taiwan, for pre-clinical work for a compound against various forms
of cancer. Dr. Alice Yu, a member of our board of directors, is a Distinguished Research Fellow
and Associate Director at the Genomics Research Center, Academia Sinica. In connection with the
agreement, we are required to pay Academia Sinica approximately $0.2 million over the term of the
agreement.
Effective March 5, 2010, the Frost Group assigned two license agreements with Academia Sinica
to the Company. The license agreements pertain to alpha-galactosyl ceramide analogs and their use
as immunotherapies and peptide ligands in the diagnosis and treatment of cancer. In connection
with the assignment of the two licenses, the Company agreed to reimburse the Frost Group for the
licensing fees previously paid by the Frost Group to Academia Sinica in the amounts of $50 thousand
and $75 thousand, respectively, as well as reimbursement of certain expenses of $50 thousand.
Effective September 21, 2009, we entered into an agreement pursuant to which we invested $2.5
million in Cocrystal in exchange for 1,701,723 shares of Cocrystals Convertible Series A Preferred
Stock. A group of Investors, led by the Frost Group (the CoCrystal Investors), previously
invested $5 million in Cocrystal, and agreed to invest an additional $5 million payable in two
equal installments in September 2009 and March 2010. As a result of an amendment to the CoCrystal
Investors agreements dated June 9, 2009, OPKO, rather than the CoCrystal Investors, made the first
installment investment ($2.5 million) on September 21, 2009. Refer to Note 2.
On September 18, 2009, we entered into the Preferred Purchase Agreement with various
investors. Refer to Note 6. Included among the investors is the Gamma Trust, Hsu Gamma
Investment, L.P, a limited partnership controlled by Jane H. Hsiao and Oracle Partners LP, a
limited partnership in which Dr. Frost is a limited partner.
On July 20, 2009, we entered into a worldwide exclusive license agreement with Academia Sinica
in Taipei, Taiwan, for a new technology to develop protein vaccines against influenza and other
viral infections. Dr. Alice Yu, a member of our board of directors, is a Distinguished Research
Fellow and Associate Director at the Genomics Research Center, Academia Sinica.
On June 16, 2009, we entered into an agreement to lease approximately 10,000 square feet of
space in Hialeah, Florida to house manufacturing and service operations for our ophthalmic
instrumentation business (the Hialeah Facility) from an entity controlled by Dr. Frost and Dr.
Jane Hsiao. Pursuant to the terms of a lease agreement, which is effective as of February 1, 2009,
gross rent is $0.1 million per year for a one-year lease and was extended through February 1, 2011.
From April 2008 through January 2009, we leased 20,000 square feet at the Hialeah Facility from a
third party landlord pursuant to a lease agreement which contained an option to purchase the
facility. We initially elected to exercise the option to purchase the Hialeah Facility in
September 2008. Prior to closing, however, we assigned the right to purchase the Hialeah Facility
to an entity controlled by Drs. Frost and Hsiao and leased a smaller portion of the facility as a
result of several factors, including our inability to obtain outside financing for the purchase,
current business needs, the reduced operating costs for the smaller space, and the minimization of
risk and expense of unutilized space.
78
Table of Contents
On June 10, 2009, we entered into a stock purchase agreement with Sorrento, pursuant to which
we invested $2.3 million in Sorrento. Refer to Note 2. In exchange for the investment, we
acquired approximately one-third of the outstanding common shares of Sorrento and received a
fully-paid, exclusive license to the Sorrento antibody library for the discovery and development of
therapeutic antibodies in the field of ophthalmology. On September 21, 2009, Sorrento entered into
a merger transaction with Quikbyte Software, Inc. Prior to the merger transaction, certain
investors, including Dr. Frost and other members of OPKO
management, made an investment in Quikbyte. Dr. Richard Lerner, a member of our Board of Directors, serves as a
consultant and scientific advisory board member to Sorrento and owns less than one percent of its
shares.
On May 26, 2009, May 29, 2009, and June 1, 2009, we entered into stock purchase agreements
with a total of seven accredited investors pursuant to which we agreed to sell an aggregate of 31
million shares of the Companys Common Stock in exchange for $31 million. Under the terms of each
investment, OPKO issued shares to the investors at a price of $1.00 per share. Refer to Note 6.
Oracle Partners, LP and Vector Group Ltd. were among the investors in the transaction and purchased
4 million and 5 million shares of our Common Stock, respectively. At the time of the investment,
Dr. Frost may also be deemed to beneficially own 11.5% of Vector Group Ltd.s outstanding stock.
On March 4, 2009, the Gamma Trust advanced $3.0 million to us under a Promissory Note we
issued to the Gamma Trust, which was repaid in full on April 27, 2009, including interest of $48
thousand. Refer to Note 5.
In March 2009, we paid the $45 thousand filing fee to the Federal Trade Commission in
connection with filings made by us and Dr. Frost, under the Hart-Scott-Rodino Antitrust
Improvements Act of 1976 (HSR). The filings permitted Dr. Frost and his affiliates to acquire
additional shares of our Common Stock upon expiration of the HSR waiting period on March 23, 2009.
On February 23, 2009, we entered into a Stock Purchase Agreement with the Gamma Trust, of
which Dr Frost is the sole trustee. Refer to Note 6.
On September 10, 2008, in exchange for a $15.0 million cash investment in the Company, we
issued 13,513,514 shares of our Common Stock to a group of investors which included members of the
Frost Group. The shares were issued at a price of $1.11 per share, representing an approximately
40% discount to the 5 day average trading price of our stock on the NYSE Amex. Refer to Note 6.
In November 2007, we entered into an office lease with Frost Real Estate Holdings, LLC, an
entity affiliated with Dr. Frost. The lease is for approximately 8,300 square feet of space in an
office building in Miami, Florida, where the Companys principal executive offices are located. We
had previously been leasing this space from Frost Real Estate Holdings on a month-to-month basis
while the parties were negotiating the lease. The lease provides for payments of approximately $18
thousand per month in the first year increasing annually to $24 thousand per month in the fifth
year, plus applicable sales tax. The rent is inclusive of operating expenses, property taxes and
parking. The rent for the first year was reduced to reflect a $30 thousand credit for the costs of
tenant improvements. From January 1, 2008 through October 1, 2008, we leased an additional 1,100
square feet of general office and laboratory space on a ground floor annex of our corporate office
building pursuant to an addendum to the Lease, which required us to pay annual rent of $19 thousand
per year for the annex space.
On September 19, 2007, we entered into an exclusive technology license agreement with Winston
Laboratories, Inc. (Winston). On February 23, 2010, we provided Winston notice of termination of
the license agreement, and the agreement terminated on May 24, 2010. Previously, members of the
Frost Group beneficially owned approximately 30% of Winston Pharmaceuticals, Inc., and Dr.
Uppaluri, our Chief Financial Officer, served as a member of Winstons board. Effective May 19,
2010, the members of the Frost Group sold 100% of Winstons capital stock beneficially owned by
them (consisting of an aggregate of 18,399,271 outstanding shares of common stock and warrants to
purchase an aggregate of 8,958,975 shares of common stock) to an entity whose members include Dr.
Joel E. Bernstein, the President and Chief Executive Officer of Winston. As consideration for the
sale, the Frost Group members received an aggregate of $789,500 in cash and non-recourse promissory
notes in the aggregate principal amount of $10,263,500 (the Promissory Notes). Dr. Uppaluri
resigned from the Winston board effective May 19, 2010. In connection with the license agreement,
we reimbursed Winston $29 thousand, and $3 thousand in the years ended December 31, 2009 and 2008,
respectively, for services provided by Winston personnel to assist us with the clinical program for
the product we licensed.
As part of the merger with Acuity Pharmaceuticals, Inc. (Acuity) in 2007, we assumed a line
of credit with the Frost Group from Acuity and amended and restated that line of credit to increase
borrowing availability. In
79
Table of Contents
connection with the increase of the borrowing availability, we issued 4,000,000 warrants to
the Frost Group. Refer to Note 5.
We reimburse Dr. Frost for Company-related use by Dr. Frost and our other executives of an
airplane owned by a company that is beneficially owned by Dr. Frost. We reimburse Dr. Frost in an
amount equal to the cost of a first class airline ticket between the travel cities for each
executive, including Dr. Frost, traveling on the airplane for Company-related business. We do not
reimburse Dr. Frost for personal use of the airplane by Dr. Frost or any other executive; nor do we
pay for any other fixed or variable operating costs of the airplane. For the fiscal years ending
December 31, 2010, 2009, and 2008, we reimbursed Dr. Frost approximately $46 thousand, $92
thousand, and $108 thousand, respectively, for Company-related travel by Dr. Frost and other OPKO
executives.
During the year ending December 31, 2008, we reimbursed SafeStitch Medical, Inc.
(SafeStitch) approximately $49 thousand for time SafeStitchs personnel spent assisting us with
the implementation of certain quality and control standard operating procedures at our
manufacturing facility in Toronto, Ontario. Dr. Hsiao serves as chairman of the board of directors
for SafeStitch; Steven Rubin and Richard Pfenniger, each of whom are members of our board of
directors, also serve on the board of directors of SafeStitch. We have not reimbursed SafeStitch
any amounts in 2010 or 2009.
Note 12 Employee Benefit Plans
Effective January 1, 2007, the OPKO Health Savings and Retirement Plan (Plan) permits
employees to contribute up to 50% of qualified pre-tax annual compensation up to annual statutory
limitations. The discretionary company match for employee contributions to the Plan is 100% of up
to the first 4% of the participants earnings contributed to the Plan. Our matching contributions
to the plan were approximately $0.2 million for each of the years ended December 31, 2010 and 2009.
Note 13 Commitments and Contingencies
On January 7, 2010, we received a letter from counsel to Nidek Co., Ltd. (Nidek) alleging
that Ophthalmic Technologies, Inc. (OTI) or OPKO breached its service obligations to Nidek under
the Service Agreement between OTI, Nidek and Newport Corporation, dated December 29, 2006, and the
Service Agreement by and between Nidek and OTI, dated the same date. We have had discussions with
Nidek regarding the matter, but it is too early to assess the likelihood of litigation in this
matter or the probability of a favorable or unfavorable outcome. We do not believe this matter
will have a material impact on our results of operations or financial condition. We are also
assessing possible claims of indemnification against a supplier in connection with the matter.
On May 6, 2008, we completed the acquisition of Vidus. Pursuant to a Securities Purchase
Agreement with Vidus, each of its stockholders, and the holders of convertible promissory notes
issued by Vidus, we acquired all of the outstanding stock and convertible debt of Vidus in exchange
for (i) the issuance and delivery at closing of 658,080 shares of our Common Stock (the Closing
Shares); (ii) the issuance of 488,420 shares of our Common Stock to be held in escrow pending the
occurrence of certain development milestones (the Milestone Shares); and (iii) the issuance of
options to acquire 200,000 shares of our Common Stock. Additionally, in the event that the stock
price for our Common Stock at the time of receipt of approval or clearance by the U.S. Food & Drug
Administration of a pre-market notification 510(k) relating to the Aquashunt is not at or above a
specified price, we will be obligated to issue an additional 413,850 shares of our Common Stock.
We are a party to other litigation in the ordinary course of business. We do not believe that
any such litigation will have a material adverse effect on our business, financial condition, or
results of operations.
We expect to incur substantial losses as we continue the development of our product
candidates, continue our other research and development activities, and establish a sales and
marketing infrastructure in anticipation of the commercialization of our diagnostic and
pharmaceutical product candidates. We currently have limited commercialization capabilities, and
it is possible that we may never successfully commercialize any of our diagnostic and
pharmaceutical product candidates. We do not currently generate revenue from any of our diagnostic
and pharmaceutical product candidates. Our research and development activities are budgeted to
expand over a period of time and will require further resources if we are to be successful. As a
result, we believe that our operating losses are likely to be substantial over the next several
years. We will need to obtain additional funds to further develop our research and development
programs, and there can be no assurance that additional capital will be available to us on
acceptable terms, or at all.
80
Table of Contents
Note 14 Strategic Alliances
We plan to develop a portfolio of product candidates through a combination of internal
development and external partnerships. On December 10, 2010, we entered into a definitive
agreement granting TESARO exclusive rights to the development, manufacture, commercialization and
distribution of rolapitant and a related compound. Refer to Note 2. We have also completed
strategic deals with the Trustees of the University of Pennsylvania, the University of Florida
Research Foundation, the University of Texas Southwestern, and Academia Sinica, among others. In
connection with these license agreements, upon the achievement of certain milestones we are
obligated to make certain payments and upon sales of products developed under the license
agreements, have royalty obligations. At this time, we are unable to estimate the timing and
amounts of payments as the obligations are based on future development of the licensed products.
Note 15 Leases
We conduct certain of our operations under operating lease agreements. Rent expense was
approximately $1.0 million for the year ended December 31, 2010, and $0.7 million for the year
ended December 31, 2009.
As of December 31, 2010, the aggregate future minimum lease payments under all non-cancelable
operating leases with initial or remaining lease terms in excess of one year are as follows:
| Year Ending | (in thousands) | |||
2011 |
$ | 506 | ||
2012 |
270 | |||
2013 |
| |||
2014 |
| |||
2015 |
| |||
Total minimum lease commitments |
$ | 776 | ||
Note 16 Segments
We currently manage our operations in two reportable segments, pharmaceutical and
instrumentation segments. The pharmaceutical segment consists of two operating segments, our (i)
pharmaceutical research and development segment which is focused on the research and development of
pharmaceutical products, diagnostic tests and vaccines, and (ii) the pharmaceutical operations we
acquired in Chile and Mexico through the acquisition of OPKO Chile and Exakta-OPKO. The
instrumentation segment consists of ophthalmic instrumentation products and the activities related
to the research, development, manufacture and commercialization of those products. There are no
inter-segment sales. We evaluate the performance of each segment based on operating profit or
loss. There is no inter-segment allocation of interest expense and income taxes.
81
Table of Contents
Information regarding our operations and assets for the two segments and the unallocated
corporate operations as well as geographic information are as follows:
| For the years ended December 31, | ||||||||||||
| (in thousands) | 2010 | 2009 | 2008 | |||||||||
Product revenue |
||||||||||||
Pharmaceutical |
$ | 21,763 | $ | 4,418 | $ | | ||||||
Instrumentation |
8,386 | 8,729 | 9,440 | |||||||||
Corporate |
| | | |||||||||
| $ | 30,149 | $ | 13,147 | $ | 9,440 | |||||||
Operating income (loss) |
||||||||||||
Pharmaceutical |
$ | 373 | $ | (11,920 | ) | $ | (19,437 | ) | ||||
Instrumentation |
(5,971 | ) | (6,843 | ) | (9,704 | ) | ||||||
Corporate |
(11,631 | ) | (9,257 | ) | (9,422 | ) | ||||||
| $ | (17,229 | ) | $ | (28,020 | ) | $ | (38,563 | ) | ||||
Depreciation and amortization |
||||||||||||
Pharmaceutical |
$ | 2,082 | $ | 507 | $ | 29 | ||||||
Instrumentation |
1,673 | 1,797 | 1,753 | |||||||||
Corporate |
115 | 53 | 41 | |||||||||
| $ | 3,870 | $ | 2,357 | $ | 1,823 | |||||||
Net loss of investees |
||||||||||||
Pharmaceutical |
$ | (714 | ) | $ | (353 | ) | $ | | ||||
Instrumentation |
| | | |||||||||
Corporate |
| | | |||||||||
| $ | (714 | ) | $ | (353 | ) | $ | | |||||
Product revenue |
||||||||||||
United States |
$ | 827 | $ | 813 | $ | 112 | ||||||
Chile |
17,977 | 4,418 | | |||||||||
Mexico |
4,110 | 24 | 1 | |||||||||
All others |
7,235 | 7,892 | 9,327 | |||||||||
| $ | 30,149 | $ | 13,147 | $ | 9,440 | |||||||
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
Assets |
||||||||
Pharmaceutical |
$ | 51,599 | $ | 28,813 | ||||
Instrumentation |
8,637 | 12,262 | ||||||
Corporate |
17,610 | 46,355 | ||||||
| $ | 77,846 | $ | 87,430 | |||||
During the year ended December 31, 2010, we also recorded $6.7 million of license revenue
related to our license agreement with TESARO and is part of our pharmaceutical business.
During the year ended December 31, 2010, one customer represented 13% of our total product
revenue. During the year ended December 31, 2009, no customers represented greater than 10% of
revenue. During the year ended December 31, 2008, four customers represented 18%, 17%, 13%, and
11%, respectively, of revenue. As of December 31, 2010, two customers represented 32% and 11% of
our accounts receivable balance. As of December 31, 2009, two customers represented 32% and 19% of
our accounts receivable balance.
Note 17 Fair Value Measurement
We record fair value at an exit price, representing the amount that would be received to sell
an asset or paid to transfer a liability in an orderly transaction between market participants. As
such, fair value is a market-based measurement that should be determined based on assumptions that
market participants would use in pricing an asset or liability. We utilize a three-tier fair value
hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include: Level
1, defined as observable inputs such as quoted prices in active markets; Level 2, defined as inputs
other than quoted prices in active markets that are either directly or indirectly observable; and
Level 3, defined as unobservable inputs in which little or no market data exists, therefore
requiring an entity to develop its own assumptions.
82
Table of Contents
As of December 31, 2010, we held money market funds that qualify as cash equivalents and
forward contracts for inventory purchases (Refer to Note 18) that are required to be measured at
fair value on a recurring basis. Our other assets and liabilities carrying value approximate their
fair value due to their short-term nature.
Any future fluctuation in fair value related to these instruments that is judged to be
temporary, including any recoveries of previous write-downs, would be recorded in accumulated other
comprehensive income or loss. If we determine that any future valuation adjustment was
other-than-temporary, we would record a charge to the consolidated statement of operations as
appropriate.
Our financial assets and liabilities measured at fair value on a recurring basis, are as
follows:
| Fair value measurements as of December 31, 2010 | ||||||||||||||||
| Quoted prices | ||||||||||||||||
| in active | Significant | |||||||||||||||
| markets for | other | Significant | ||||||||||||||
| identical | observable | unobservable | ||||||||||||||
| assets | inputs | inputs | ||||||||||||||
| (in thousands) | (Level 1) | (Level 2) | (Level 3 | Total | ||||||||||||
Assets: |
||||||||||||||||
Money market funds |
$ | 16,895 | $ | | $ | | $ | 16,885 | ||||||||
Liabilities: |
||||||||||||||||
Forward contracts |
$ | | $ | 689 | $ | | $ | 689 | ||||||||
Note 18 Derivative Contracts
We enter into foreign currency forward exchange contracts to cover the risk of exposure to
exchange rate differences arising from inventory purchases on letters of credit. Under these
forward contracts, for any rate above or below the fixed rate, we receive or pay the difference
between the spot rate and the fixed rate for the given amount at the settlement date.
We record derivative financial instruments on our balance sheet at their fair value as an
accrued expense and the changes in the fair value are recognized in income in other expense net
when they occur, the only exception being derivatives that qualify as hedges. To qualify the
derivative instrument as a hedge, we are required to meet strict hedge effectiveness and
contemporaneous documentation requirements at the initiation of the hedge and assess the hedge
effectiveness on an ongoing basis over the life of the hedge. At December 31, 2010, the forward
contracts did not meet the documentation requirements to be designated as hedges. Accordingly, we
recognize all changes in fair values in income.
The outstanding contracts at the end of the year 2010 have been valued at fair value, and
their maturity details are as follows:
| (in thousands) | Fair value at | |||||||||||
| Days until maturity | Contract value | December 31, 2010 | Effect on loss | |||||||||
0 to 30 |
$ | 359 | $ | 386 | $ | (27 | ) | |||||
31 to 60 |
1,129 | 1,244 | (115 | ) | ||||||||
61 to 90 |
1,924 | 2,061 | (137 | ) | ||||||||
91 to 120 |
2,787 | 3,033 | (246 | ) | ||||||||
121 to 180 |
1,192 | 1,335 | (143 | ) | ||||||||
More than 180 |
379 | 400 | (21 | ) | ||||||||
Total |
$ | 7,770 | $ | 8,459 | $ | (689 | ) | |||||
83
Table of Contents
Note 19 Selected Quarterly Financial Data (Unaudited)
| For the 2010 Quarters Ended | ||||||||||||||||
| (in thousands) | March 31 | June 30 | September 30 | December 31 | ||||||||||||
Revenue |
$ | 7,922 | $ | 7,455 | $ | 7,599 | $ | 13,904 | ||||||||
Gross margin |
2,394 | 2,605 | 2,344 | 9,036 | ||||||||||||
Net loss attributable to
common shareholders |
(5,346 | ) | (6,876 | ) | (8,010 | ) | (1,318 | ) | ||||||||
Basic and diluted loss
per share |
$ | (0.02 | ) | $ | (0.03 | ) | $ | (0.03 | ) | $ | (0.01 | ) | ||||
| For the 2009 Quarters Ended | ||||||||||||||||
| (in thousands) | March 31 | June 30 | September 30 | December 31 | ||||||||||||
Revenue |
$ | 2,301 | $ | 2,347 | $ | 1,501 | $ | 6,998 | ||||||||
Gross (deficit) margin |
740 | 583 | 446 | 1,811 | ||||||||||||
Net (loss) income attributable to
common shareholders |
(9,055 | ) | (5,734 | ) | (10,298 | ) | (9,744 | ) | ||||||||
Basic and diluted (loss) income
per share |
$ | (0.05 | ) | $ | (0.03 | ) | $ | (0.04 | ) | $ | (0.04 | ) | ||||
Due to rounding, the quarterly per share amounts may not mathematically compute to the annual
amount.
On December 10, 2010, we licensed our rolapitant development program and as a result, recorded
$6.7 million of revenue. Refer to Note 2. In addition, we acquired Exakta-OPKO on February 16,
2010. On October 7, 2009 we acquired OPKO Chile. The results of operations include the results of
Exakta-OPKO and OPKO Chile after their acquisitions. Refer to Note 2. In the fourth quarter of
2009, we recorded a $1.1 million impairment charge related to goodwill associated with our
instrumentation business. Refer to Note 2. In the quarter ended September 30, 2009, we recorded a
$3.9 million preferred stock dividend related to a beneficial conversion feature of our Series D
Preferred Stock. Refer to Note 6.
Note 20 Subsequent Events
On March 14, 2011, we issued 27,000,000 shares of our Common Stock in a public offering at a
price of $3.75 per share. The net proceeds received were approximately $96.4 million after
deducting the underwriters discounts and commissions and other estimated offering expenses. We
also granted the underwriters a 30-day option to purchase up to an additional 4,050,000 shares of
our Common Stock to cover overallotments, if any. On March 15, 2011, representatives for the underwriters provided us notice that the underwriters
exercised a portion of their 4,050,000 share over-allotment option for 2,397,029 additional shares of our Common
Stock. As part of the offering, Frost Gamma Investments
Trust, of which Phillip Frost is the sole trustee, and Hsu Gamma Investment, L.P., of which Jane
Hsiao, the Companys Vice Chairman and Chief Technical Officer, serves as the general partner,
purchased an aggregate of 3,733,000 shares of our Common Stock at the
public offering price. Jefferies & Company, Inc. and J.P. Morgan
Securities LLC acted as joint book-running managers for the offering.
UBS Investment Bank and Lazard Capital Markets LLC acted as co-lead
managers for the offering and Ladenburg Thalmann & Co. Inc., a
subsidiary of Ladenburg Thalmann Financial Services Inc., acted as
co-manager for the offering. Dr.
Frost is the Chairman of the Board of Directors and principal shareholder of Ladenburg Thalmann
Financial Services Inc.
On February 22, 2011, we entered into Amendment No. 2 (the Amendment) to our Credit
Agreement, dated March 27, 2007, as amended, with the Frost Group (the Credit Agreement). The
Amendment renewed the Companys $12.0 million line of credit with the Frost Group. The line of
credit, which previously expired on January 11, 2011, was renewed until March 31, 2012 on
substantially the same terms as in effect at the time of expiration. We are obligated to pay
interest upon maturity, compounded quarterly, on outstanding borrowings under the line of credit at
an 11% annual rate.
On January 28, 2011, we entered into a definitive agreement (the CURNA Merger Agreement)
with CURNA, Inc., (CURNA) and each of CURNAs shareholders and optionholders, pursuant to which
we agreed to acquire all of the outstanding stock of CURNA in exchange for $10 million in cash.
CURNA was a privately held company based in Jupiter, Florida, engaged in the discovery of new
drugs for the treatment of a wide variety of illnesses, including cancer, heart disease, metabolic
disorders and a range of genetic anomalies. Closing of the transaction occurred on January 31,
2011.
We have reviewed all subsequent events and transactions that occurred after the date of our
December 31, 2010 consolidated balance sheet date, through the time of filing this Annual Report on
Form 10-K.
84
Table of Contents
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
ITEM 9A. CONTROLS AND PROCEDURES.
Disclosure Controls and Procedures
The Companys management, under the supervision and with the participation of the Companys
Chief Executive Officer (CEO) and Chief Financial Officer (CFO), has evaluated the
effectiveness of the Companys disclosure controls and procedures as defined in Securities and
Exchange Commission (SEC) Rule 13a-15(e) as of December 31, 2010. Based on that evaluation, CEO
and CFO have concluded that the Companys disclosure controls and procedures are effective to
ensure that information the Company is required to disclose in reports that it files or submits
under the Securities Exchange Act is accumulated and communicated to management, including the CEO
and CFO, as appropriate, to allow timely decisions regarding required disclosure and is recorded,
processed, summarized, and reported within the time periods specified in the SECs rules and forms.
Managements Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over
financial reporting, as such term is defined in Rule 13a-15(f) under the Securities Exchange Act of
1934, as amended. Our internal control over financial reporting is a process designed to provide
reasonable assurance regarding the reliability of financial reporting and the preparation of
financial statements according to generally accepted accounting principles. All internal control
systems, no matter how well designed, have inherent limitations. Therefore, even those systems
determined effective could provide only reasonable assurance with respect to financial statement
preparation and presentation.
Our management conducted an evaluation of the effectiveness of our internal control over
financial reporting as of December 31, 2010, based on the framework in Internal Control -
Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway
Commission.
Based on our evaluation under the framework in Internal Control Integrated Framework,
management concluded that our internal control over financial reporting was effective as of
December 31, 2010.
The effectiveness of the Companys internal control over financial reporting as of December
31, 2010 has been audited by Ernst & Young LLP, our independent registered public accounting firm,
who also audited our consolidated financial statements included in this Annual Report on Form 10-K,
as stated in their report which appears with our accompanying consolidated financial statements.
Changes to the Companys Internal Control Over Financial Reporting
As part of the Companys September 30, 2010 close process, the Company identified that it had
not properly accounted for a beneficial conversion feature on, and the classification of
convertible preferred stock. As a result, the Company has implemented additional controls and
procedures over financial reporting including adding additional review procedures on its complex
accounting issues. In addition, in connection with our acquisitions of Exakta-OPKO and OPKO Chile,
we continue to implement a new accounting system, as well as standards and procedures, upgrading
and establishing controls over accounting systems and adding employees who are trained and
experienced in the preparation of financial statements in accordance with U.S. GAAP to ensure that
we have appropriate internal control over financial reporting at Exakta-OPKO and OPKO Chile. Other
than as set forth above with respect to Exakta-OPKO and OPKO Chile and the additional review
procedures of complex accounting issues, there have been no changes to the Companys internal
control over financial reporting that occurred during the Companys fourth fiscal quarter that have
materially affected, or are reasonably likely to materially affect, the Companys internal control
over financial reporting.
ITEM 9B. OTHER INFORMATION.
None.
85
Table of Contents
PART III
The information required in Items 10 (Directors, Executive Officers and Corporate Governance),
Item 11 (Executive Compensation), Item 12 (Security Ownership of Certain Beneficial Owners and
Management and Related Stockholder Matters), Item 13 (Certain Relationships and Related
Transactions, and Director Independence), and Item 14 (Principal Accounting Fees and Services) is
incorporated by reference to the Companys definitive proxy statement for the 2011 Annual Meeting
of Stockholders to be filed with the Securities and Exchange Commission within 120 days of December
31, 2010.
86
Table of Contents
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES.
| (a) (1) | Financial Statements: See Part II, Item 8 of this report. |
| (2) | We filed our consolidated financial statements in Item 8 of Part II. Additionally, the financial statement schedule entitled Schedule II Valuation and Qualifying Accounts has been omitted since the information required is included in the consolidated financial statements and notes thereto. |
| (3) | Exhibits: See below. |
| Exhibit | ||||
| Number | Description | |||
| 2.1(1) | Merger Agreement and Plan of Reorganization, dated as of March 27,
2007, by and among Acuity Pharmaceuticals, Inc., Froptix Corporation,
eXegenics, Inc., e-Acquisition Company I-A, LLC, and e-Acquisition
Company II-B, LLC. |
|||
| 2.2(5)+ | Securities Purchase Agreement dated May 6, 2008, among Vidus Ocular,
Inc., OPKO Instrumentation, LLC, OPKO Health, Inc., and the
individual sellers and noteholders named therein. |
|||
| 2.3(11) | Purchase Agreement, dated February 17, 2010, among Ignacio Levy
García and José de Jesús Levy García, Inmobiliaria Chapalita, S.A. de
C.V., Pharmacos Exakta, S.A. de C.V., OPKO Health, Inc., OPKO Health
Mexicana S. de R.L. de C.V., and OPKO Manufacturing Facilities S. de
R.L. de C.V. |
|||
| 3.1(2) | Amended and Restated Certificate of Incorporation. |
|||
| 3.2(4) | Amended and Restated By-Laws. |
|||
| 3.3(9) | Certificate of Designation of Series D Preferred Stock. |
|||
| 4.1(1) | Form of Common Stock Warrant. |
|||
| 4.2(9) | Form of Common Stock Warrant. |
|||
| 10.1(1) | Form of Lockup Agreement. |
|||
| 10.2(1) | License Agreement, dated as of March 31, 2003, by and between the
Trustees of the University of Pennsylvania and Acuity
Pharmaceuticals, Inc. (Reich/Tolentino). |
|||
| 10.3(1) | License Agreement, dated as of March 31, 2003, by and between the
Trustees of the University of Pennsylvania and Acuity
Pharmaceuticals, Inc. (Reich/Gewirtz). |
|||
| 10.4(1) | First Amendment to License Agreement, dated as of August 1, 2003, by
and between the Trustees of the University of Pennsylvania and Acuity
Pharmaceuticals, Inc. (Reich/Tolentino). |
|||
| 10.5(1) | First Amendment to License Agreement, dated as of August 1, 2003, by
and between the Trustees of the University of Pennsylvania and Acuity
Pharmaceuticals, Inc. (Gewirtz). |
|||
| 10.6(1) | Credit Agreement, dated as of March 27, 2007, by and among eXegenics,
Inc., The Frost Group, LLC, and Acuity Pharmaceuticals, LLC. |
|||
| 10.7(1) | Amended and Restated Subordination Agreement, dated as of March 27,
2007, by and among The Frost Group, LLC, Horizon Technology Funding
Company LLC, Acuity Pharmaceuticals, LLC, and eXegenics, Inc. |
|||
| 10.8(4) | Share Purchase Agreement, dated April 11, 2007, by and between
Ophthalmic Technologies, Inc. and eXegenics, Inc. |
|||
87
Table of Contents
| Exhibit | ||||
| Number | Description | |||
| 10.9(3) | Lease Agreement dated November 13, 2007, by and between Frost Real
Estate Holdings, LLC and the Company. |
|||
| 10.10(4) | Share Purchase Agreement, dated as of November 28, 2007, by and among
Ophthalmic Technologies, Inc., OTI Holdings Limited, and the
Shareholders named therein. |
|||
| 10.11(4) | Exchange and Support Agreement, dated as of November 28, 2007, by and
among OPKO Health, Inc. and OTI Holdings Limited and the holders of
exchangeable shares named therein. |
|||
| 10.12(4) | Stock Purchase Agreement, dated December 4, 2007, by and between
members of The Frost Group, LLC and the Company. |
|||
| 10.13(4)* | OPKO Health, Inc. 2007 Equity Incentive Plan. |
|||
| 10.14(5) | Form of Director Indemnification Agreement. |
|||
| 10.15(5) | Form of Officer Indemnification Agreement. |
|||
| 10.16(6) | Stock Purchase Agreement, dated August 8, 2008 by and among the
Company and the Investors named therein. |
|||
| 10.17(7) | Stock Purchase Agreement, dated February 23, 2009 by and between the
Company and Frost Gamma Investments Trust. |
|||
| 10.18(7) | Promissory Note to Frost Gamma Investments Trust, dated March 4, 2009. |
|||
| 10.19(8) | Form of Stock Purchase Agreement for transactions between the Company
and Nora Real Estate SA., Vector Group Ltd., Oracle Partners LP,
Oracle Institutional Partners, LP., Chung Chia Company Limited, Gold
Sino Assets Limited and Grandtime Associates Limited. |
|||
| 10.20(8) | Stock Purchase Agreement, dated June 10, 2009, by and among the
Company and Sorrento Therapeutics, Inc. |
|||
| 10.21(9) | Form of Securities Purchase Agreement Series D Preferred Stock. |
|||
| 10.22(10)* | Form of Restricted Share Award Agreement (Director). |
|||
| 10.23(10) | Cocrystal Discovery, Inc. Agreements. |
|||
| 10.24(13) | Stock Purchase Agreement, dated October 1, 2009, by and among the
OPKO Chile Limitada and Inversones OPKO Limitada, subsidiaries of the
Company, and the Sellers named therein. |
|||
| 10.25+(12) | Asset Purchase Agreement, dated October 12, 2009, by and between the
Company and Schering Corporation. |
|||
| 10.26(12) | Letter Agreement, dated June 29, 2010, by and between the Company and
Schering Corporation. |
|||
| 10.27+ | Exclusive License Agreement by and between the Company and TESARO,
Inc. dated December 10, 2010. |
|||
| 21 | Subsidiaries of the Company. |
|||
| 23.1 | Consent of Ernst & Young LLP. |
|||
| 31.1 | Certification by Phillip Frost, Chief Executive Officer, pursuant to
Exchange Act Rules 13a-14 and 15d-14. |
|||
88
Table of Contents
| Exhibit | ||||
| Number | Description | |||
| 31.2 | Certification by Rao Uppaluri, Chief Financial Officer, pursuant to
Exchange Act Rules 13a-14 and 15d-14. |
|||
| 32.1 | Certification by Phillip Frost, Chief Executive Officer, pursuant to
18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002. |
|||
| 32.2 | Certification by Rao Uppaluri, Chief Financial Officer, pursuant to
18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the
Sarbanes-Oxley Act of 2002. |
|||
| * | Denotes management contract or compensatory plan or arrangement. | |
| + | Certain confidential material contained in the document has been omitted and filed separately with the Securities and Exchange Commission. | |
| (1) | Filed with the Companys Current Report on Form 8-K filed with the Securities and Exchange Commission on April 2, 2007, and incorporated herein by reference. | |
| (2) | Filed with the Companys Current Report on Form 8-A filed with the Securities and Exchange Commission on June 11, 2007, and incorporated herein by reference. | |
| (3) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 14, 2007 for the Companys three-month period ended September 30, 2007, and incorporated herein by reference. | |
| (4) | Filed with the Companys Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2008 and incorporated herein by reference. | |
| (5) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 8, 2008 for the Companys three-month period ended June 30, 2008, and incorporated herein by reference. | |
| (6) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 12, 2008 for the Companys three-month period ended September 30, 2008, and incorporated herein by reference. | |
| (7) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 8, 2009 for the Companys three-month period ended March 31, 2009, and incorporated herein by reference. | |
| (8) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 7, 2009 for the Companys three-month period ended June 30, 2009, and incorporated herein by reference. | |
| (9) | Filed with the Companys Current Report on Form 8-K filed with the Securities and Exchange Commission on September 24, 2009, and incorporated herein by reference. | |
| (10) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 9, 2009 for the Companys three-month period ended September 30, 2009, and incorporated herein by reference. | |
| (11) | Filed with the Companys Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 10, 2010 for the Companys three-month period ended March 31, 2010, and incorporated herein by reference. | |
| (12) | Filed with the Companys Amendment to Annual Report on Form 10-K filed with the Securities and Exchange Commission on February 3, 2011. | |
| (13) | Filed with the Companys Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 17, 2010. |
89
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934,
the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized.
| OPKO HEALTH, INC. | ||||||
| By: | /s/ Phillip Frost, M.D.
|
|||||
| Phillip Frost, M.D. | ||||||
| Chairman of the Board and | ||||||
| Chief Executive Officer | ||||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been
signed below by the following persons on behalf of the registrant and in the capacities and on the
dates indicated.
| Signature | Title | Date | ||
/s/ Dr. Phillip Frost, M.D.
|
Chairman of the Board and Chief Executive Officer (Principal Executive Officer) |
March 16, 2011 | ||
/s/ Dr. Jane H. Hsiao
|
Vice Chairman and Chief Technical Officer | March 16, 2011 | ||
/s/ Steven D. Rubin
|
Director and Executive Vice President Administration |
March 16, 2011 | ||
/s/ Rao Uppaluri
|
Senior Vice President and Chief
Financial Officer (Principal Financial Officer) |
March 16, 2011 | ||
/s/ Adam Logal
|
Executive Director of Finance, Chief
Accounting Officer and Treasurer (Principal Accounting Officer) |
March 16, 2011 | ||
/s/ Robert Baron
|
Director | March 16, 2011 | ||
/s/ Thomas E. Beier
|
Director | March 16, 2011 | ||
/s/ Pascal J. Goldschmidt, M.D.
|
Director | March 16, 2011 | ||
/s/ Richard A. Lerner, M.D.
|
Director | March 16, 2011 | ||
/s/ John A. Paganelli
|
Director | March 16, 2011 | ||
/s/ Richard C. Pfenniger, Jr.
|
Director | March 16, 2011 | ||
/s/ Alice Lin-Tsing Yu, M.D., Ph.D.
|
Director | March 16, 2011 |
90
Table of Contents
EXHIBIT INDEX
| Exhibit | ||||
| Number | Description | |||
| 10.27+ | Exclusive License Agreement by and between
the Company and TESARO, Inc. dated December
10, 2010. |
|||
| 21 | Subsidiaries of the Company. |
|||
| 23.1 | Consent of Ernst & Young LLP. |
|||
| 31.1 | Certification by Phillip Frost, Chief
Executive Officer, pursuant to Exchange Act
Rules 13a-14 and 15d-14. |
|||
| 31.2 | Certification by Rao Uppaluri, Chief
Financial Officer, pursuant to Exchange Act
Rules 13a-14 and 15d-14. |
|||
| 32.1 | Certification by Phillip Frost, Chief
Executive Officer, pursuant to 18 U.S.C.
Section 1350, as adopted pursuant to Section
906 of the Sarbanes-Oxley Act of 2002. |
|||
| 32.2 | Certification by Rao Uppaluri, Chief
Financial Officer, pursuant to 18 U.S.C.
Section 1350, as adopted pursuant to Section
906 of the Sarbanes-Oxley Act of 2002. |
|||
| + | Certain confidential material contained in the document has been omitted and filed separately with the Securities and Exchange Commission. |
91